Nature子刊发重磅综述介绍肿瘤的诺奖方向(值得收藏)
解螺旋公众号·陪伴你科研的第2580天
癌症治疗方向
2018年诺贝尔生理学或医学奖授予两位免疫学家:美国的詹姆斯·艾利森与日本的庶佑,以表彰他们“发现负性免疫调节治疗癌症的疗法方面的贡献”。这几年对关于免疫治疗的机制研究也是大家关注的焦点。所以,无论我们只是想要蹭蹭,还是想要深入地研究免疫治疗相关的分子机制,
一篇回顾了免疫治疗相关分子机制的高质量综述就成了我们的宝藏!

2020年5月28日,中国科学院上海生物化学与细胞生物学研究所的许琛琦教授在著名期刊Cell Research上发表了题为《Immune checkpoint signaling and cancer immunotherapy》的综述性文章,为我们系统性的介绍了免疫检查点信号和肿瘤免疫。许教授课题组在T细胞的基础研究中发现了T细胞活化的新分子机制,同时开拓了代谢调控这一肿瘤免疫治疗的新领域,发展了新的肿瘤免疫治疗方法。妥妥一枚大佬!
为了帮助大家更好的吸收综述的内容,榨干其每一滴精华,本工在阅读综述的时候,也给大家翻译了遍,希望能给大家带来有真正价值的“学术营养”。

摘要
免疫检查点阻断疗法已成为治疗癌症的主要武器。相关抗体药物,如抗PD-1和抗PD-L1,显示出了明显的优势,包括可广泛适用于各种癌症类型,以及当治疗有效时可产生持久的临床反应。然而,其总体应答率仍然不令人满意,特别是对于突变负荷较低的肿瘤。此外,不良反应,如自身免疫症状和肿瘤超进展(hyperprogression),在一些临床应用中呈现出明显的不利影响。这些挑战反映了全面了解免疫检查点生物学特征的迫切需要。
在这篇综述中,我们讨论了免疫检查点信号在多个水平上的调节,并提供相关概述来阐述我们目前对检查点生物学的理解。本文讨论的主题包括通过表面传递、内化、循环和降解来调节已知免疫检查点蛋白的表面表达水平。当到达表面时,检查点与配体可进行常规的反式和顺式相互作用,以诱导信号和调节免疫反应。除了经典的检查点阻断之外,最近也已经出现了靶向这些途径的新的治疗策略,并在临床前模型中进行了测试,为开发下一代免疫疗法提供了新的方向。
引言
肿瘤微环境(TME)中浸润着多种天然免疫细胞和获得性免疫细胞,其免疫监视功能往往受到多种机制的抑制。
信号抑制和代谢抑制是免疫抑制的两个主要原因
,在本文中将讨论
前者
(代谢抑制在本公众号之前的推送中27分的Cell子刊再出重磅综述,给你的国自然加油有相关介绍)。信号抑制反映在肿瘤细胞下调刺激性免疫受体的活性,而上调抑制性免疫受体的活性。以T细胞为例,肿瘤细胞可以通过下调表面MHC-I的表达水平来调节T细胞受体(TCR)介导的刺激信号。另一方面,肿瘤细胞可以通过上调表面PD-L1水平来调节PD-1介导的抑制信号。阻断抑制性免疫受体的激活可以恢复免疫细胞的抗肿瘤功能,这一概念已经在实验中得到证实,并已在临床上应用于多种癌症的治疗。
在过去的几十年里,许多抑制性免疫受体在肿瘤中被发现和研究,
包括但不限于
PD-1
、
CTLA-4
、
LAG3
、
TIM3
、
TIGIT
和
BTLA
。它们被称为“
免疫检查点
”,指的是充当免疫反应守门人的分子。在进化过程中,免疫检查点与刺激性免疫受体共同进化,早在鱼类中就出现了。这些受体通常包含单酪氨酸信号基序,如免疫受体酪氨酸抑制基序(ITIM)和免疫受体酪氨酸转换基序(ITSM)来传递抑制信号。作为表面分子,它们的活性很容易被阻止配体-受体结合的抗体所抑制。
最成功
的免疫检查点阻断疗法是
抗PD-1/PD-L1疗法
,该疗法已被批准用于治疗各种类型的癌症,如血液肿瘤、皮肤癌、肺癌、肝癌、膀胱癌和肾癌。免疫检查点阻断治疗通常能比化疗或靶向治疗产生更持久的反应,这可能也反映了免疫系统的记忆特征。
然而,随着临床数据在全球范围内的积累,缺点和副作用也开始暴露了出来。免疫检查点阻断治疗的主要瓶颈是其在大多数癌症中的应答率较低,范围在10%-30%之间。对于一些主要的癌症类型,如具有微卫星稳定的结直肠癌,抗PD-1/PD-L1治疗几乎没有效果。治疗无应答的机制也已被广泛研究,许多因素被发现是相关的,如肿瘤突变负荷、PD-L1表达水平、IFN信号和MHC-I缺失。然而,能够准确预测疗效的生物标记物仍然缺乏。因此,迫切需要更好地理解检查点生物学(checkpoint biology),以设计下一代治疗方法,并改进现有疗法的临床方案。
近年来,许多生化和生物物理的研究揭示了检查点表面表达的复杂调控。在与配体结合后,不同的检查点显示出不同的信号机制来抑制抗肿瘤免疫。在这里,我们回顾了这些基本的发现,并强调了具有临床转化潜力的新靶向策略。
免疫检查点的表面水平调节
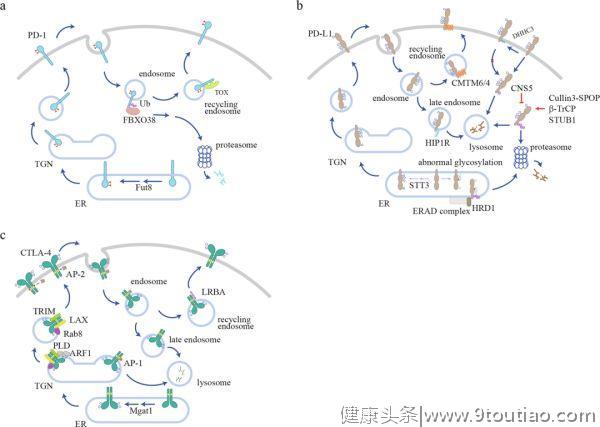
高水平的检查点是TME的一个标志,但其潜在机制尚不清楚。作为一种膜蛋白,免疫检查点在内质网(ER)中表达,然后被运送到细胞表面发挥其抑制功能,这一过程通过蛋白分选系统依次通过高尔基体和分泌囊泡进行运输。在表面传递过程中,糖基化起到质量控制的作用,以确保只有成熟的和功能正常的免疫检查点才能传递到细胞表面。到达细胞表面后,免疫检查点需要被内化和再循环,这为调节其表面水平提供了一条快速的调节途径。泛素化介导的蛋白降解是控制蛋白质水平的另一个重要机制,免疫检查点可以被泛素化并分类到蛋白酶体或溶酶体中进行降解。这些细胞过程共同决定了免疫检查点的表面水平,从而形成细胞信号(图1如下)。
1
PD-1的调控
人PD-1的胞外IGV结构域有4个N-连接的
糖基化位点
:
N49
、
N58
(小鼠PD-1的N54)、
N74
和
N116
。PD-1的结构表明,N58位的糖链由两个N’乙酰氨基葡萄糖和一个岩藻糖(fucose)组成。此外,全基因组CRISPR筛选证实,核心岩藻糖基化通路直接调节PD-1在细胞表面的水平。质谱分析表明,
4个N-糖基化位点均有核心岩藻糖修饰
。N49或N74突变导致细胞表面PD-1水平显著降低。核心岩藻糖基转移酶8(Fut8)的遗传切除(Genetic ablation)降低了PD-1的表面水平,增强了T细胞的激活。
糖基化如何调节PD-1的表达水平尚不清楚。
糖基可能会调节PD-1折叠
,从而影响内质网的质量控制过程。糖基化的
另一个可能作用是调节配体结合
。不同的微环境信号,如缺氧和营养应激,可能导致PD-1糖基化模式不同,进而影响PD-1的功能。
未来有必要进行更多的质谱研
究
,以了解PD-1的“糖码(sugar code)”及其在特定疾病背景下的功能意义。
荧光成像已经观察到了表面PD-1的内化,但尚不清楚传统的笼状蛋白所介导的内吞作用是否参与了PD-1的内化。内化的PD-1分子可以经循环(recycle)回到细胞表面,或者被泛素化修饰,并分类到蛋白酶体进行降解(图1a)。
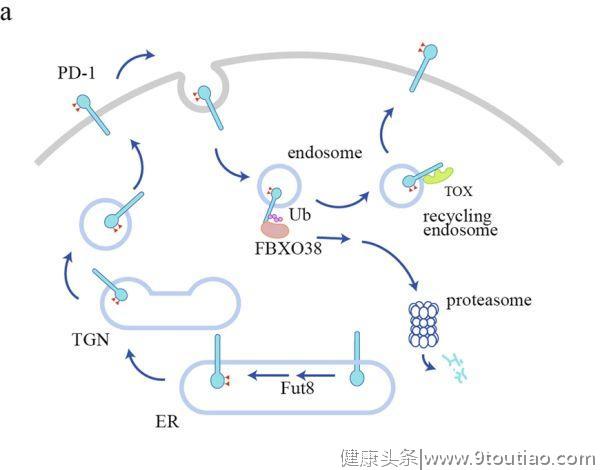
在肝癌浸润的CD8+T细胞中,胸腺细胞选择相关的高迁移率群盒蛋白(Tox)是T细胞耗竭的主要转录因子,具有与胞浆中的PD-1相结合并促进PD-1循环的非常规功能。Tox是否也在其他情况下调节PD-1的循环(recycle)还没有得到检验。
PD-1的一种特异性E3泛素连接酶,FBXO38,已被生化和动物实验证实。作为含有Skp、Cullin、F-box的复合物(SCF复合物)的一部分,FBXO38可以介导K48在保守的赖氨酸位点(人类PD-1中的K233)发生多泛素化。有趣的是,多泛素化的PD-1会被分选到蛋
白酶体而不是溶酶体
中进行降解。这一现象并不常见,因为大多数膜蛋白都是通过溶酶体内化和降解的。
在TME中,由于FBXO38转录水平较低,FBXO38所介导的PD-1降解通路存在缺陷。没有CD28信号的TCR信号被发现是FBXO38下调的原因。因此,肿瘤抗原的持续暴露和肿瘤细胞表面CD80/86的低表达可以解释TIL中FBXO38表达降低的原因。
此外,T细胞的主要生长因子IL-2可以通过STAT5介导的转录调控来恢复肿瘤浸润性T细胞中FBXO38的水平。值得注意的是,FBXO38在肿瘤浸润性淋巴细胞(TIL)中的表达水平甚至低于幼稚T细胞。
慢性TCR信号如何下调FBXO38转录
仍是一个悬而未决的问题。
事实上,总的来说,人们对PD-1分子内化和随后降解或循环过程的调控仍然知之甚少。一些有趣的问题也值得进一步研究,例如PD-1的内化是否依赖于信号,哪些信号决定了PD-1的内化,以及它是被传递到蛋白酶体进行降解,还是被循环回到细胞表面供进一步使用。
2
PD-L1的调控
PD-L1(又称CD127、B7-H1)也含有4个
N-糖基化位点
:
N35
、
N192
(小鼠PD-L1中的N191)、
N200
(小鼠PD-L1中的N199)和
N219
(小鼠PD-L1中的N218)。这些修饰对PD-L1蛋白的稳定性有重要意义。STT3是一种ER相关的N-糖基转移酶,可催化蛋白质发生N-糖基化的第一步反应。在肿瘤干细胞中,STT3依赖的N-糖基化可稳定并上调PD-L1的水平,这一过程是上皮-间充质转化(EMT)诱导PD-L1富集所必需的。相反,AMPK使PD-L1 S195发生磷酸化,导致PD-L1糖基化异常,阻止其ER向高尔基体转运,进而发生ER相关降解(ERAD)。
在某些癌细胞中,多糖修饰使得PD-L1不能被常规抗体检测到,这会导致对PD-L1表面水平产生误解。去N-糖基化可以更准确地检测PD-L1的表面水平。这一发现反映了这样一个事实,即
PD-L1的糖基化模式在不同的癌细胞中可能会有所不同
,这可能是由于它们的微环境不同,而且
其中一些模式也阻止了与常规抗体的结合
。
细胞表面PD-L1经历不断的内化,然后循环或降解(图1b)。
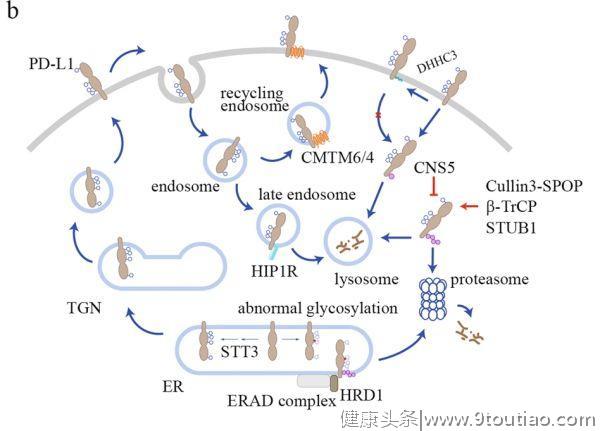
一种伴侣蛋白,CKLF样Marvel跨膜结构域6(CMTM6),属于8个包含Marvel结构域的蛋白家族之一,功能未知,可调节PD-L1的循环。CMTM6与PD-L1在质膜和内吞体(endosomes)内结合,促进回收(recycling),并抑制溶酶体对其泛素化修饰和降解。CMTM6缺乏的肿瘤细胞表现出PD-L1在循环和表面的水平降低,导致T细胞活性较少受到抑制。类似于CMTM6的CMTM4也具有类似的功能。CMTM6/4是如何支持PD-L1被回收利用的,其机制仍未确定。
目前已鉴定出多种可调节PD-L1溶酶体降解的蛋白质。HIP1R携带溶酶体分选基序,其与PD-L1结合后可通过AP复合物和Alix/ESCRT将PD-L1靶向溶酶体。提示STUB1 E3泛素连接酶可介导溶酶体中PD-L1的降解。STUB1是否会与HIP1R协作(cooperates)仍是未知。根据几个小组的研究结果,蛋白酶体也参与了PD-L1的降解。据报道,Cullin3-SPOP、β-TrCP和Hrd1E3连接酶可调节PD-L1的泛素化和蛋白酶体降解,它们似乎会在不同的环境中调节PD-L1的降解。
在细胞周期中,PD-L1的表面水平有明显的波动,在M期和G1期早期达到峰值,在G1期晚期和S期迅速下降。这种波动受细胞周期素D-CDK4-SPOP-FZR1信号通路的调节。CDK4磷酸化并稳定了SPOP,SPOP是Cullin 3-based 的 E3 泛素连接酶复合物中的衔接蛋白,介导了PD-L1的多泛素化和蛋白酶降解。有趣的是,糖基化可以通过β-TrCP和HRD1直接影响PD-L1的泛素化和降解。当PD-L1未糖基化时,可被糖原合成酶激酶3β(GSK3β)在T180和S184位点发生磷酸化,并募集β-TrCP介导PD-L1泛素化和降解。另一方面,S195的磷酸化会导致PD-L1的糖基化异常,进而募集HIP1R,从而触发ER相关的降解。
另外,也有些复杂的机制可对抗PD-L1泛素化和降解。据报道,COP9信号体5(CSN5)能去泛素化PD-L1,从而抑制PD-L1的降解。TNF-α激活的NF-κB通路可诱导肿瘤细胞表达CSN5,从而稳定PD-L1的表达。DHHC3可将PD-L1的C272位发生棕榈酰化,阻断了PD-L1的单一泛素化以及随后ESCRT介导转运到多囊泡小体(MVB),从而抑制了PD-L1在溶酶体中的降解。
3
CTLA-4调控
与主要定位在质膜的PD-1不同,
CTLA-4主要定位于细胞内
。当T细胞被激活时,CTLA-4才被转移(translocates)到细胞表面,介导其抑制功能。T细胞受体相互作用分子(TRIM)是CTLA-4从反式高尔基体网络(TGN)运输到细胞表面所必需的。敲降TRIM可导致CTLA-4保留在TGN中。随后的研究表明,CTLA-4/TRIM/LAX/Rab8复合物对这一转运途径是必不可少的。磷脂酶D(PLD)和ADP核糖化因子1(ARF1)依赖的胞吐作用也被报道可促使CTLA-4转运到细胞表面。
细胞表面CTLA-4分子可被迅速内化,以维持相对较低的表面水平(图1C)。
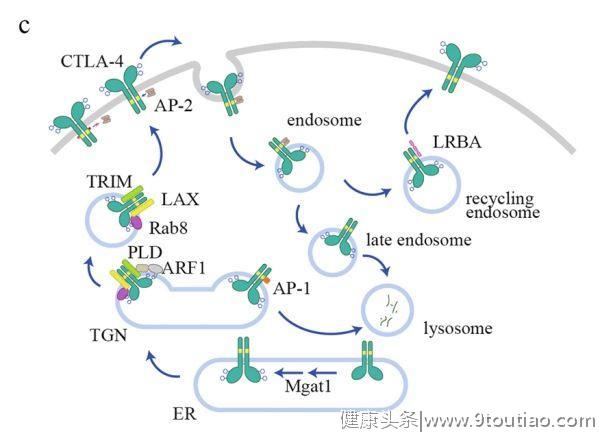
网格蛋白相关衔接复合物 AP-2可与CTLA-4细胞质结构域中的YVKM基序结合并介导内化,而YVKM磷酸化可阻碍这一过程。然而,另一项研究表明,YVKM介导的CTLA-4内化在T细胞活化过程中没有被阻碍,因此YVKM磷酸化可能不直接调节CTLA-4内化。另一个网状蛋白衔接复合物AP-1也与YVKM基序结合,但不同的是它将CTLA-4从TGN转运到溶酶体进行降解。
此外,
CTLA-4的内化率也受N-糖基化的调节
。维生素D3处理后增强了N-乙酰氨基葡萄糖基转移酶I(Mgat1)的表达和N-糖链分支,导致T细胞CTLA-4的内化减少和表面水平增加。N-糖基化也是CTLA-4表面传递所必需的。信号肽T17A的多态性可导致糖基化不足和CTLA-4表面水平降低。TCR信号可增加氨基己糖代谢和N-糖链分支通路,从而增加CTLA-4糖基化和表面表达。内吞体中的内化 CTLA-4的CTLA-4可以循环回到细胞表面。LPS反应性米色锚蛋白(LRBA)与CTLA-4共定位于内吞体循环中,协助其循环利用。患者的LRBA发生突变可降低调节性和常规T细胞中CTLA-4的水平,从而导致表现出自身免疫、淋巴增殖和体液免疫缺陷的表型。
检查点信号机制
免疫检查点的抑制功能通常依赖于配体诱导的信号转导。在这里,我们总结了几个研究得较为深入免疫检查点的配体相互作用和信号机制(图2如下)。
1
PD-1信号
PD-1信号可以通过与配体PD-L1和PD-L2的结合而被触发。一般而言,表达在抗原提呈细胞或肿瘤细胞表面的PD-L1或PD-L2与表达在T细胞表面的PD-1发生反式相互作用,从而诱导产生抑制信号。肿瘤细胞还可以分泌含有PD-L1的胞外小泡,主要以外泌体的形式激活PD-1途径。这些外泌体形式的PD-L1分子主要抑制引流淋巴结中的T细胞活性。抗PD-1治疗无效的黑色素瘤患者外泌体形式的PD-L1水平高于有反应者。
最近的研究表明,PD-1/PD-L1的相互作用也可表现为顺式。PD-L1和PD-1在APC上的共表达和相互作用阻止了PD-1的反式连接,从而降低了T细胞PD-1的抑制功能。除PD-1外,PD-L1还可与顺式细胞中的CD80相互作用,破坏PD-L1/PD-1和CD80-CTLA-4的相互作用,但保留CD80激活CD28信号的能力。因此,顺式PD-L1-CD80的相互作用可通过抑制PD-1和CTLA-4的功能以在抗肿瘤免疫中发挥积极作用。
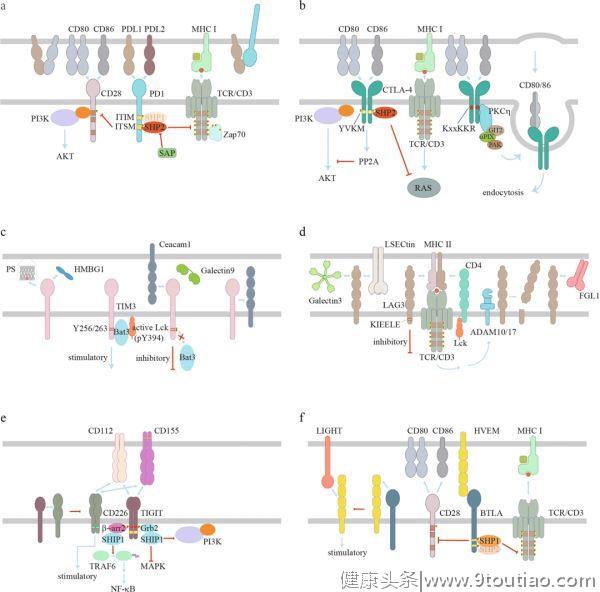
配体结合后,PD-1被磷酸化以传递其抑制功能(图2a)。
ITIM和ITSM均存在于其胞质区域。磷酸化的ITSM可能更重要,因为ITSM中的酪氨酸突变会显著地降低PD-1的抑制功能,而ITIM中的酪氨酸突变则不会。磷酸化的ITSM主要招募SHP2对关键信号分子去磷酸化,从而下调活性水平。虽然ITIM通常被认为是PD-1抑制功能中必不可少的,但最近的研究表明,ITIM在将SHP2从非活性构象转化为活性构象的过程中起着重要作用。信号淋巴细胞活化分子相关蛋白(SAP)可以阻断SHP2与其底物的相互作用,从而抑制PD-1信号转导。
虽然SHP2在大多数情况下是PD-1抑制信号所必需的,但SHP2缺陷型(Shp2-deficient)T细胞仍然会表现出功能障碍的特征,并对α-PD-1治疗有反应,这一现象提示了另一种信号机制。根据一些研究小组的报道,磷酸化的PD-1 ITSM也可以招募SHP1。最近的一项研究应用质谱法对PD-1信号小体进行了定量研究。他们发现,SHP2主要由PD-1招募,而SHP1作为储备,以补偿SHP2的丢失,或者在SHP2变得有限的情况下,这可能是PD-1高表达的慢性/休眠T细胞的情况。有趣的是,SHP1/2双基因敲除后,PD-1仍然可有效抑制原代T细胞的增殖和细胞因子的产生,提示PD-1的抑制功能仍存在未知的机制。
研究表明,
PD-1既能抑制抗原信号,又能抑制共刺激信号
。在激活的T细胞中,PD-1可转移(translocates)到免疫突触,因此与TCR和CD28非常接近。一项生化研究表明,比起TCR,SHP2对CD28有明显的偏好。事实上,CD28下游的PI3K-AKT通路被PD-1以ITSM依赖和ITIM非依赖的方式抑制。然而,PD-1信号也显示出可抑制TCR和下游信号分子如ZAP70的磷酸化。对早期T细胞活化过程中PD-1所调控的基因的表达进行转录组分析,结果发现PD-1主要抑制强TCR信号所诱导的基因表达。与TCR相比,磷酸化的PD-1 ITSM所募集的SHP2可能更倾向于CD28,但仍能抑制TCR信号转导。除了对T细胞信号的抑制作用外,SHP2还被报道可通过逆转CSK介导的 LCK 抑制性磷酸化来激活TCR信号。有证据表明,磷酸化的PD-1可隔离(sequestration)SHP2来阻碍SHP2激活LCK活性,从而有助于抑制T细胞信号。
2
CTLA-4信号
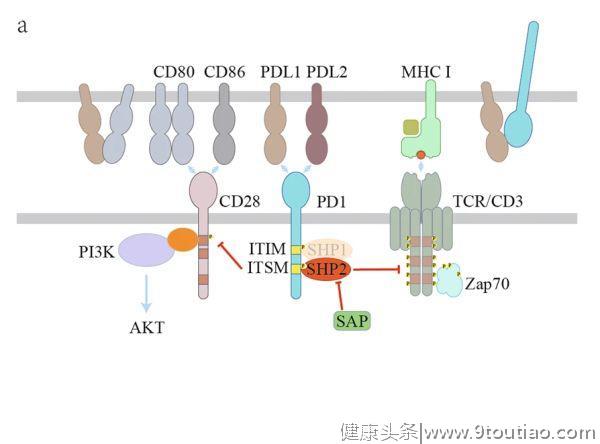
与CD28相比,CTLA-4以更高的亲和力与CD80/86结合,从而通过
配体竞争
来抑制共刺激作用。另外,表达CTLA-4的T细胞可通过反式内吞作用(trans-endocytosis)降低APC表面CD80/86的表达,从而导致CD28信号降低。例如,具有组成性CTLA-4表达的调节性T(Treg)细胞可以通过这种反式内吞过程介导树突状细胞(DC)上CD80/86的下调,这是Treg细胞发挥抑制功能所必需的。如上所述,APC表面的顺式CD80/PD-L1异源二聚体可保护CD80免受CTLA-4介导的反式内吞作用。尽管PD-L1和CD80之间的顺式相互作用破坏了PD-1和CTLA-4的抑制功能,但肿瘤细胞的CD80表达往往较低,因此这种保护机制可能无效。
当T细胞激活时,CTLA-4转移到细胞表面,并聚集到免疫突触中。CTLA-4的YVKM基序中的酪氨酸可以被Src家族激酶或其他激酶磷酸化,如JAK2和Rlk(图2b)。
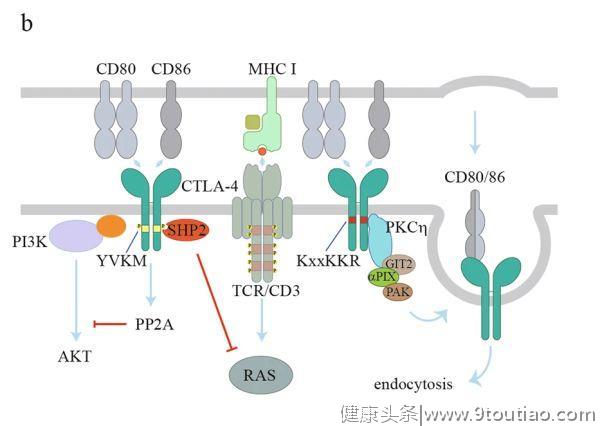
酪氨酸磷酸化可阻碍CTLA-4与AP-2之间的相互作用,从而维持CTLA-4在细胞表面传递抑制信号。另一方面,YVKM基序也可能招募SHP2以抑制T细胞活化。此外,尽管CTLA-4对PP2A的直接募集作用仍有争议,但CTLA-4对AKT活性的抑制是依赖于PP2A的。在Treg细胞中,蛋白激酶Cη(PKCη)被CTLA-4募集到免疫突触。CTLA-4/PKCη进一步募集GIT2-αPIX-PAK复合物,促进Treg-APC的相互作用,是Treg细胞的接触依赖性抑制作用所必需的。
除了胞浆尾部所介导对T细胞反应的抑制,CTLA-4还被认为以外在(extrinsic)方式抑制T细胞信号。例如,CTLA-4通过如上所述的反式内吞作用或通过诱导TGFβ下调CD80/86的表达,从而减少APC上CD80/86的表达。CTLA-4还通过连接CD80/86诱导DC表达吲哚胺2,3-双加氧酶(IDO),导致色氨酸耗竭和T细胞抑制。
3
TIM3信号
目前已报道了TIM3的四种配体,即
C型凝集素galectin9
、
癌胚抗原细胞粘附分子1
(Ceacam1)、
HMGB1
和
非蛋白配体磷脂酰丝氨酸
(PS)(图2c)。
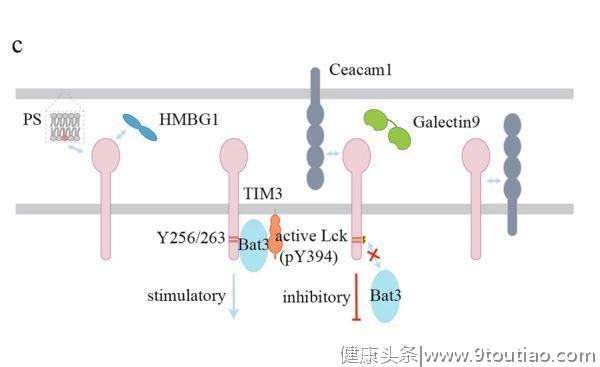
Galectin9是一种具有两个碳水化合物识别结构域的可溶性蛋白。Galectin9与TIM3的结合需要TIM3糖基化的IGV结构域。Ceacam1以顺式和反式这两种方式结合TIM3。与Ceacam1的共表达和顺式相互作用是TIM3糖基化和细胞表面表达所必需的,反式相互作用则介导抑制效应T细胞功能。
另外两种配体主要调节先天性免疫反应。HMGB1是一种可分泌到TME的非组蛋白染色质相关蛋白。HMGB1与肿瘤相关树突状细胞上的TIM3结合,抑制死亡肿瘤细胞所释放的核酸向树突状细胞内吞体募集,从而抑制核酸诱导的先天性免疫反应。此外,TIM3通过与PS直接结合来作用于胞吐作用所识别的凋亡细胞,从而调节DC的胞吐作用。TIM3抗体可抑制CD8+DC吞噬凋亡细胞,从而减少抗原交叉提呈。
另外,TIM3信号仍然存在争议,因为不同的研究小组已经报道了TIM3对T细胞效应功能的相反作用。在小鼠急性淋巴细胞性脉络膜脑膜炎病毒(LCMV)感染模型中,TIM3的表达促进了短寿命效应T细胞的发育,并伴随着AKT/mTOR信号的增强。
另一项研究表明,TIM3与免疫突触中的多个近端TCR信号分子相互作用,阻断TIM3可促进高TIM3表达的 CD8 T细胞与靶细胞之间形成稳定的突触。TIM3的胞浆结构域含有5个保守的酪氨酸残基,其中Y265(小鼠为Y256)和Y272(小鼠为Y263)可被Src家族激酶或IL-2诱导的T细胞激酶(ITK)磷酸化。一旦磷酸化,这些酪氨酸残基可以招募P85来促进NFAT的激活。
BAT3对TIM3诱导的Th1细胞死和亡耗竭具有抑制作用。当BAT3与非磷酸化的TIM3胞浆结构域结合时,BAT3可特异性地招募具有催化活性形式的Lck来促进TCR信号。TIM3与抗体或配体结合后,可能通过Y265和Y272的磷酸化导致BAT3的解离,并逆转BAT3对TIM3功能的抑制作用。因此,尽管TIM3本身可能作为抑制性受体发挥作用,但它与BAT3的相互作用(association)在某些情况下会将其转化为刺激性受体。
4
LAG3信号
LAG3被确认为是MHC-II的配体,其亲和力高于CD4,因此可能通过阻碍CD4-MHC-II的相互作用来抑制CD4+T细胞的激活。然而,其他研究表明,LAG3的抑制功能并不依赖于CD4的竞争,而是依赖于它的
胞质结构域
来传递抑制信号。然而,LAG3在CD8+T细胞中的抑制功能不涉及MHC-II,这提示LAG3可能存在其他配体。事实上,LSECtin和Gelectin-3与LAG3结合并抑制TME中的T细胞功能(图2d)。
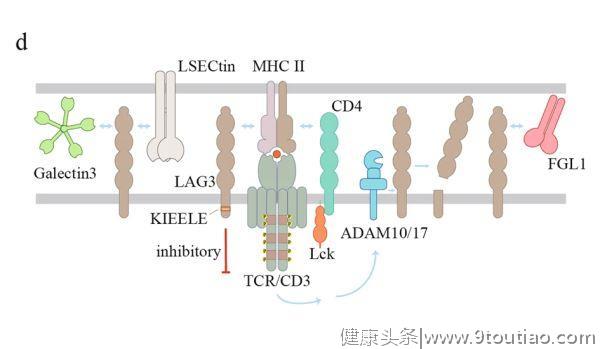
LAG3是一种糖蛋白,在胞外区有四个潜在的N-连接糖基化位点。考虑到LSECtin和Gelectin-3都是碳水化合物结合蛋白,它们与LAG3的结合可能依赖于LAG3的糖基化。最近,纤维蛋白原样蛋白1(FGL1)被鉴定为LAG3的新配体。正常情况下,FGL1从肝脏中以低水平释放到血液中。
然而,在几种人类癌症中都检测到了上调的FGL1。阻断FGL1和LAG3之间的相互作用可增强T细胞的抗肿瘤功能。有趣的是,LAG3也在Tregs中表达,以抑制其增殖和功能。同时,Treg表达的LAG3与APC上的MHC-II连接络合(ligation)后也会抑制APC的功能。因此,LAG3的作用是复杂的,将LAG3阻断用于癌症免疫治疗需要进一步仔细研究后才能确认临床益处。
对LAG3信号转导的认识仍然有限。CD3和LAG3的交联可抑制T细胞的增殖和细胞因子的产生,这可能是由于近端TCR信号被削弱(impairing)和钙内流减少而导致的。LAG3的胞质结构域在人和小鼠中都含有三个保守区,即丝氨酸磷酸化位点、KIEELE基序和多个EP重复序列。
KIEELE序列对于LAG3在CD4+T细胞中的抑制功能是必不可少的。通过两种跨膜金属蛋白酶(A去整合素和金属蛋白酶结构域蛋白10和17(ADAM10和ADAM17))裂解LAG3,LAG3的功能会被TCR信号所拮抗。TCR信号可通过不同的机制上调ADAM10和ADAM17的切割活性,进而促进T细胞的增殖和功能。
5
TIGIT信号
CD155(PVR)
和
CD112(PVRL2)
是TIGIT的两个配体,其亲和力高于 CD122。TIGIT的反式连接不仅会通过TIGIT信号途径传递T细胞和NK细胞的抑制性信号,而且可通过逆向(reverse)CD155信号来促进DCs产生IL-10从而抑制T细胞功能。CD226是一种共刺激受体,与TIGIT具有相同的配体。而TIGIT与其配体的亲和力高于CD226,因此TIGIT可通过配体竞争抑制CD226介导的共刺激作用。有趣的是,TIGIT也可以顺式的方式直接结合CD226来阻碍其同二聚体的形成和共刺激功能。
关于TIGIT信号的研究主要集中在NK细胞中。TIGIT的胞质结构域包含一个ITIM基序和一个免疫球蛋白尾部酪氨酸(ITT)样基序(图2e)。
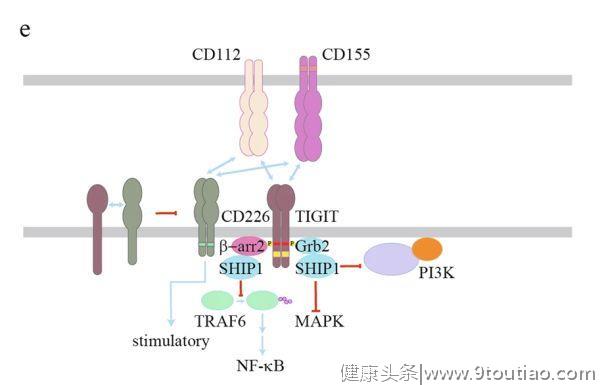
不同的研究表明,无论是ITIM基序还是ITT样基序,酪氨酸磷酸化都是TIGIT抑制NK细胞功能的重要因素。然而,在鼠类的NK细胞中,这两个基序似乎是多余的。据报道,ITT样结构域可通过Grb2和β-arrestin2两种衔接蛋白招募SHIP1。Grb2招募的SHIP1主要抑制PI3K和MAPK信号,而β-arrestin招募的SHIP1主要抑制TRAF6以降低NF-κB的活性。然而,TIGIT中ITIM基序的下游信号尚不清楚。
6
BTLA信号
BTLA和CD160共享相同的配体,即疱疹病毒进入介质(HVEM),以抑制T细胞功能。然而,当HVEM与TNF超家族成员LIGHT或BTLA/CD160结合时,HVEM本身就能传递共刺激信号。BTLA/CD160和LIGHT可与HVEM的不同位点结合,例如可与富含半胱氨酸的结构域1 (CRD1) 区域相互作用。然而,HVEM的CRD1截断并不影响LIGHT的结合。有趣的是,可溶性LIGHT增强了BTLA/HVEM的相互作用,而据报道,膜相关的LIGHT可取代BTLA,因为它对HVEM的亲和力更高。当BTLA和HVEM共表达时会发生顺式相互作用,这会阻碍HVEM被反式连接(ligation)所激活。
BTLA在其胞质结构域中含有ITIM和ITSM序列,以及Grb2识别序列。发生连接后,ITIM和ITSM中的酪氨酸残基均可被磷酸化,并诱导SHP1/SHP2抑制T细胞功能(图2f)。
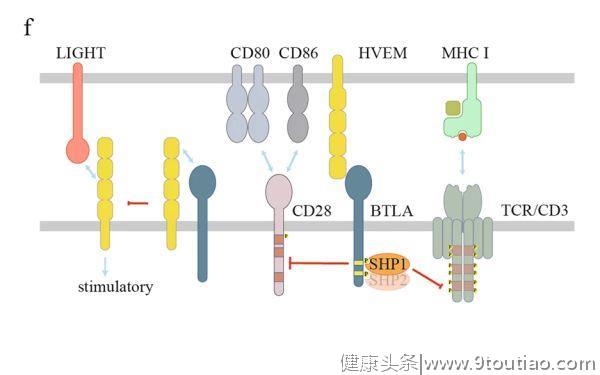
进一步比较BTLA和PD-1之间的信号,与PD-1招募更弱的磷酸酶 SHP2相反,BTLA更倾向于招募更有效的磷酸酶 SHP1,以更有效地抑制TCR和CD28信号。此外,当HVEM在B细胞表面表达时,T滤泡辅助细胞(Tfh)上的BTLA可将SHP1招募到免疫突触中,从而抑制TCR信号,抑制CD40L以抑制B细胞增殖。
靶向免疫检查点表达的治疗策略
使用抗体阻断受体-配体相互作用的免疫检查点阻断疗法已被批准应用于临床。然而,这些阻断抗体的总体反应率仍然很低。鉴于免疫检查点的抑制功能受到其表面表达和信号转导的严格调节,靶向这些通路可以为免疫疗法提供新的策略(表1)。
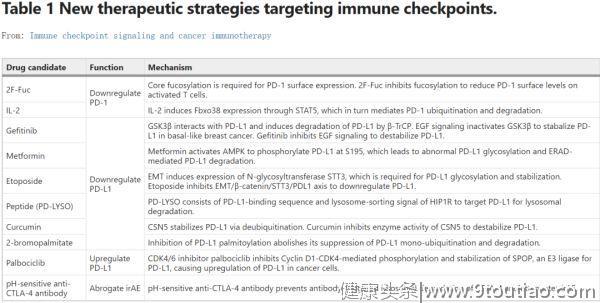
许多开创性研究探索了靶向检查点糖基化和泛素化/降解途径的可能性。这些实验是在不同的系统中进行的,在这里我们将它们列在一起,以突出这种新方法的转化潜力:
(1)
靶向检查点糖化
。检查点在细胞表面的稳定表达需要适当的糖基化。用岩藻糖基化抑制剂2F-Fuc处理T细胞,可降低PD-1的岩藻糖基化和表面水平。2F-Fuc处理的细胞毒性T淋巴细胞(CTL)在过继细胞转移(ACT)治疗期间表现出抗肿瘤免疫活性增强。
PD-L1的糖基化受 AMPK 和EMT调节。二甲双胍是一种广泛用于治疗2型糖尿病的药物,它能激活AMPK以诱导PD-L1的异常糖基化和降解。依托泊苷是一种化学疗法类药物,可用于治疗各种癌症,可抑制由EMT诱导的PD-L1糖基化,破坏细胞表面PD-L1的稳定性。二甲双胍和依托泊苷可下调肿瘤细胞表面PD-L1的表达,增强抗CTLA-4和抗TIM治疗的疗效。
PD-L1糖基化也受EGFR信号调节。吉非替尼是一种EGFR抑制剂,已用于治疗多种癌症,也能抑制PD-L1糖基化,进而促进GSK3β介导的泛素化和降解,从而提高抗PD-1治疗的疗效。
(2)
靶向检查点泛素化/降解
。促进检查点的降解似乎是一个有意思的方向。IL-2,一种FDA 批准的治疗转移性黑色素瘤和肾癌的药物,可上调FBXO38介导的PD-1泛素化/降解。这可能是IL-2治疗癌症的几种分子机制之一。
一种设计出来的肽PD-LYSO,包含有PD-L1结合序列和来自HIP1R的溶酶体分选信号序列,可以靶向PD-L1进行溶酶体降解。姜黄素通过抑制CSN5的去泛素化活性以破坏PD-L1的稳定性,从而有利于抗CTLA-4的治疗。
如上所述,PD-L1的棕榈酰化可以抑制单泛素化和降解,进而稳定表面表达。2-溴棕榈酸酯(2-bromopalmitate)可抑制PD-L1棕榈酰化以降低PD-L1的表面水平,以在鼠MC38肿瘤模型中促进抗肿瘤免疫。
另一方面,在某些情况下,提高PD-L1的表面水平也被证明是有益的。CDK4/6抑制剂palbociclib可抑制细胞周期蛋白D-CDK4-SPOP-FZR1通路介导的PD-L1泛素化和降解,增加PD-L1水平并使植入CT26的肿瘤对抗PD-1治疗敏感。
最后,CTLA-4阻断抗体用于癌症免疫治疗通常会引起严重的免疫治疗相关的不良反应(irAEs)。最近的一项研究表明,容易发生irAE的CTLA-4阻断抗体可诱导CTLA-4的溶酶体降解,而不易发生irAE的抗体则允许CTLA-4以LBRA依赖性方式循环/回收。提高有irAE倾向的抗CTLA-4抗体对pH的敏感性可以防止抗体引起的CTLA-4溶酶体降解并降低irAE。
观点
免疫检查点的抑制功能受到表面表达水平、受体-配体相互作用和细胞内信号转导的严格调控。目前的免疫检查点阻断疗法旨在靶向受体-配体相互作用。除了这种已成功的方法,最近的研究表明,调节表面表达和细胞内信号也可能是抗肿瘤免疫的新希望。尽管在该领域已取得了令人兴奋的进展,但未来的研究仍有待解决几个主题,以为下一代免疫疗法铺平道路:
(1)免疫检查点的翻译后修饰(PTMs)。目前的研究强调了糖基化、脂质修饰和泛素化在检查点功能中的重要性。然而,我们对检查点PTM的理解仍然非常有限。为了系统地研究免疫检查点的修饰,我们需要先进的质谱测定技术。
(2)免疫检查点的周转(Turn-over)过程。作为膜蛋白,免疫检查点的表面表达水平由多个细胞生物学过程所控制,包括表面传递、内化、回收和降解。到目前为止,人们对这些过程知之甚少,因此需要确定所涉及的关键调控蛋白。
(3)免疫检查点的细胞内信号机制。大多数检查点需要酪氨酸磷酸化激活抑制信号,但是对磷酸化过程还没有深入的研究。此外,检查点磷酸化后所募集的效应分子也没有得到很好的表征。事实上,不同的检查点更倾向于不同的效应分子来执行其功能。例如,PD-1主要招募SHP2,BTLA主要招募SHP1。这些不同特性的潜在意义尚不清楚。SHP2和SHP1是否在免疫抑制中发挥不同的作用也不完全清楚。因此,还需要进行更多的实验来填补这些空白。
(4)免疫检查点的背景依赖性(Context-dependent)生物学特征。最近的研究结果表明,免疫检查点受特定的调节机制的影响,并在不同的免疫和癌细胞环境下表现出不同的功能。除了癌细胞,肿瘤微环境中还充满了其他各种类型的细胞,包括T细胞、巨噬细胞、中性粒细胞、树突状细胞、髓源性抑制细胞、NK细胞和癌相关的成纤维细胞。
对基础和转化研究人员来说,进一步了解TME内不同细胞类型的检查点生物学也是非常有意思和挑战的工作。例如,PD-1在效应、调节和记忆T细胞中的作用是复杂和多因素的。事实上,PD-1的阻断会导致Treg过度活动,进而导致免疫抑制而不是免疫重新激活,正如在一些接受PD-1阻断疗法的黑色素瘤患者中观察到的超进展性疾病所反映的那样。
总之,基于检查点生物学的免疫疗法代表了癌症治疗的光明前景。进一步提升我们对免疫检查点生物学的理解,将提高检查点阻断疗法的疗效,并且为将新的免疫疗法应用于临床提供重要信息。