第118期:神经内分泌-代谢功能障碍和神经退行性疾病中的睡眠
本期由姝儿分享 Eduardo Spinedi等人2018年发表在Neyro Endocrinology上的题为“Neuroendocrine-Metabolic Dysfunction and Sleep Disturbances in Neurodegenerative Disorders: Focus on Alzheimer ́s Disease and Melatonin”的文章。
摘要
阿尔茨海默病(AD)与饮食行为改变和代谢紊乱有关。在AD大脑的许多下丘脑核中观察到淀粉样斑块和神经丝缠结。其中一些区域(视交叉上核,下丘脑外侧区)也在睡眠/觉醒周期的调节中起作用,并可能解释在AD患者中观察到的进食和睡眠障碍的共病。睡眠不足会增加神经退行性疾病的进程,例如,慢波睡眠的减少会损害脑间质液中β-淀粉样肽(Aβ)和tau蛋白的清除。脑脊液(CSF)褪黑素水平即使在临床前阶段(Braak-1阶段)患者未出现认知障碍时也会降低,这表明脑脊液中褪黑素的减少可能是早期标志的迎面而来的AD。
在AD的转基因动物模型中,褪黑激素的给药增加了Aβ的淋巴清除,并减少了Aβ的产生和沉积。它也可能在下丘脑摄食信号之间建立新的平衡。虽然在AD的临床阶段进行的褪黑激素试验未能显示或仅显示出对认知的适度积极作用,但在痴呆的临床前阶段(最小认知障碍,褪黑激素的作用是显而易见的,可显著改善睡眠和生活质量。AD下丘脑改变的主要方面,睡眠中断和神经退行性变之间的联系,以及褪黑素对这些过程的可能治疗作用。
简介
神经退行性疾病是以大脑结构和功能逐渐恶化为特征的疾病。选择性神经元群体的退化在AD 和额颞叶痴呆中引起显著的认知症状,或者在帕金森病、肌萎缩性侧索硬化或亨廷顿病中引起主要的运动症状。除了神经元丢失和大脑炎症的习惯性存在之外,神经退行性疾病还与进食行为改变(食欲减退/厌食)和代谢变化(如体重减轻(瘦素血症))有关,表明下丘脑神经内分泌区域参与进食行为。大约50%-60%的AD患者表现出进食障碍 ,14%-80%的患者表现出营养不良,20%-45%的AD患者的临床特征是体重减轻。AD的主要症状是与语言缺陷、人格障碍和感觉-运动关联功能改变相关的记忆丧失 。
最常见的AD是迟发性AD,这是一种与遗传因素和环境风险因素相关的多因素、异质性疾病,而家族性AD(占病例的不到2% -3%与淀粉样前体蛋白、早老素1和早老素2的遗传突变有关。从神经病理学的角度来看,AD是由淀粉样斑块中β-淀粉样肽(Aβ)的细胞外积累和过度磷酸化tau蛋白的纤维聚集体形成的神经丝缠结(NFTs)的存在定义的 。脑组织中Aβ和tau的含量取决于与慢波睡眠相关的清除机制 。 AD下丘脑改变的主要方面,睡眠中断和神经退行性变之间的联系,以及褪黑素对这些过程的可能治疗作用。
AD的主要神经内分泌代谢功能障碍
下丘脑是一个关键的大脑结构,参与调节饮食行为的两个主要途径:食欲刺激途径(食欲增加途径)和食欲抑制途径(食欲减少途径)。位于下丘脑弓状核(ARC)的神经元合成促食欲肽神经肽Y (NPY)和agouti基因相关肽(AgRP)。它们的轴突与位于下丘脑外侧区(LHA)的二级神经元形成突触,这些细胞含有主要的促食欲肽食欲素和黑色素浓缩激素(MCH)。ARC还包含编码饱腹感的神经元,该神经元产生源自前黑皮质素(POMC)的异常肽,该异常肽与含可卡因和苯丙胺调节转录物(CART)的神经元共存。这些厌食神经元投射到下丘脑室旁核(PVN)的神经元,并增加促肾上腺皮质激素释放激素(CRH)的合成,这是另一种强有力的厌食肽。
外周信号通过受体介导的过程在下丘脑水平相互作用,以刺激或抑制食欲/厌食途径。例如,白色脂肪组织来源的瘦素和胰腺β细胞来源的胰岛素以及一些肠道来源的肽[例如胆囊收缩素、胰高血糖素样肽1、肽YY]的循环水平的提高抑制了下丘脑促食欲通路。相反,胃来源的Ghrelin,唯一的肠道来源的促食欲激素,关闭了下丘脑促食欲神经元的反馈机制 。这种机制是个体饮食行为复杂性的基础;事实上,大多数下丘脑的改变都会导致不良的代谢结果(图1)。几项研究支持AD患者的显著下丘脑萎缩。在AD患者大脑的许多下丘脑核中观察到淀粉样斑块和神经营养因子,包括PVN、LHA以及结节小脑核、视上核和视交叉上核 (图1)。其中一些区域(LHA,斯德哥尔摩)在睡眠/觉醒周期的调节中发挥作用,并可能解释在AD患者中出现的饮食和睡眠障碍的共病。肾上腺、甲状腺和性腺分泌在AD患者中也是功能障碍,这种功能障碍被认为参与了AD的生理病理学。
然而,在AD患者中,功能失调的下丘脑导致能量稳态紊乱和随之而来的代谢障碍。在改变的代谢途径中,肥胖(体重指数高于30)、胰岛素抵抗(胰岛素抵抗;有缺陷的下游胰岛素受体、胰岛素信号系统),2型糖尿病(空腹血糖等于/高于7 mM和/或葡萄糖耐量受损)和病毒感染增加了患AD的风险。AD患者的体重减轻似乎与淀粉样蛋白负荷和疾病进展有关。重要的是,大约10年后,体重减轻先于AD症状的出现。共识是下丘脑斑块和缠结存在于AD的早期-中度阶段,体重减轻通常发生在认知障碍之前。
此外,老年体重指数下降可能表明患AD的高风险和更高的AD进展率。虽然代谢障碍似乎与下丘脑功能障碍有关,但相关的信号通路尚未完全了解。除了tau和Aβ下丘脑积聚外,许多其他因素也可导致AD患者的代谢障碍。研究支持瘦素信号参与AD的能量稳态变化,例如Aβ肽改变ARC NPY神经元对瘦素的反应。事实上,尽管在转基因AD小鼠中没有观察到ARC瘦素受体基因表达的改变,但个体的低瘦素血症特征与ARC POMC和CART基因水平的降低共存,因此表明厌食途径的正常功能;相反,ARC NPY和AgRP基因在野生型和AD小鼠中的表达相似,因此表明功能障碍的ARC仅限于NPY-AgRP神经元。这一事实在禁食AD小鼠后得到了类似的注意,并得到了电生理学研究的有力支持。

具体来说,在不同的AD实验模型中,弧形病变有利于AD样病变的发展]。AD患者的体重减轻也可能是由感觉(如味觉/嗅觉)整合或加工缺陷引起的]。令人信服的证据支持葡萄糖稳态改变与AD生理病理学之间的相互关系,肥胖、胰岛素抵抗和糖尿病是AD的强风险因素。在AD转基因模型中,肥胖和胰岛素抵抗的发展恶化了淀粉样变性或τ病理学(图2)。 研究表明,脑胰岛素抵抗足以促进tau病理和淀粉样变。
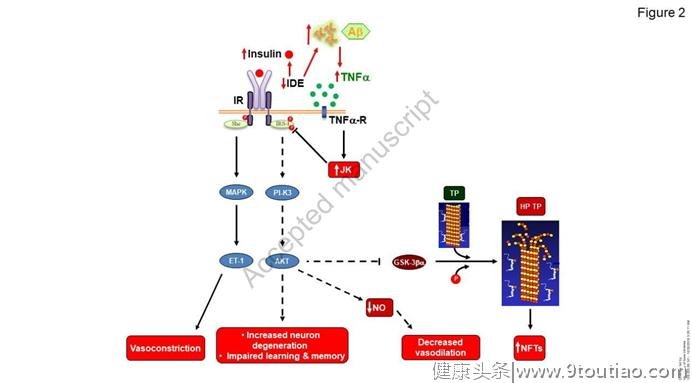
AD患者患糖尿病的风险增加,从而支持了AD中脑损伤和代谢紊乱之间的相互关系。AD大脑被称为胰岛素抵抗,这是一种与个人认知得分相关的状态。这一观察结果与胰岛素信号传导促进可塑性和记忆的已知能力一致,这可能与大脑皮层的变化或鼻内胰岛素治疗后观察到的人类记忆改善有关。胰岛素信号在能量代谢调节中的已知作用也支持了大脑胰岛素抵抗在AD患者葡萄糖稳态受损发展中的潜在作用。AD脑中胰岛素抵抗的起源似乎与Aβ和τ病理有关。小鼠侧脑室注射Aβ寡聚体通过下丘脑机制触发糖尿病前期状态(葡萄糖耐量受损),tau功能丧失损害胰岛素反应性,并与葡萄糖稳态改变相关。这与纯陶氏病患者大脑IRS-1抑制增加一致。
虽然潜在的机制尚不清楚,但已表明Aβ寡聚体可促进胰岛素受体内化以及c-Jun N末端激酶、蛋白激酶R和肿瘤坏死因子-α (TNF-α)途径的激活,进而导致IRS-1功能抑制。这些说法还得到研究的支持,研究表明,在小鼠体内icv给药Aβ寡聚体后,外周葡萄糖代谢障碍出现的时间比下丘脑炎症标记物快速增加(4小时)的时间晚了几个小时(12小时),因此表明葡萄糖代谢障碍是小鼠体内icv给药Aβ寡聚体诱导的中枢效应的结果。此外,还证明了Aβ低聚物诱导的小鼠下丘脑炎症,其特征是脑氧化应激(OS)和肿瘤坏死因子-α增强,可被抗氧化剂治疗和功能性肿瘤坏死因子-α受体缺失所覆盖。此外,最近的数据表明,在迟发性糖尿病相关的AD中,载脂蛋白E4的过量产生有助于体内胰岛素受体的捕获。总之,这些数据强调了在AD患者中发现的葡萄糖稳态损伤可能是由异常胰岛素信号引起的下丘脑损伤引起的(图2)。
AD的睡眠障碍
AD神经退行性变超越认知功能,涉及到包括进食和睡眠在内的关键生理过程。所涉及的过程可以作为生物标志物来帮助疾病的早期诊断]。在老年人中,睡眠效率降低到大约80%,睡眠开始潜伏期增加,N1和N2阶段(轻度慢睡)和睡眠开始后醒来的时间百分比增加。其他与年龄相关的下降是深度慢波睡眠(N3阶段)和快速动眼期(快速眼动)睡眠的减少。
多导睡眠图数据的脑电图功率谱分析证实,老年人的非快速眼动(NREM)睡眠和快速眼动睡眠减少,δ活动显著减少。AD患者睡眠障碍的发生率接近70 %,并且它们通常在认知恶化开始之前出现。与同龄的非痴呆个体[38]相比,AD患者的睡眠结构表明慢波睡眠和快速眼动睡眠的定量减少,以及睡眠碎片化的显著程度,这降低了白天的警觉性并增加了午睡。大约一半的AD患者在下午晚些时候/晚上早些时候表现出神经精神症状的恶化,伴有焦虑、不安和混乱(“日落”)。睡眠障碍和“日落”是这些病人住院的主要原因。
睡眠和神经退行性疾病之间的关系是双向的。神经变性伴随着睡眠困难,这是由于昼夜节律(如褪黑激素分泌)的振幅和相位变化减少,以及神经退行性过程对睡眠的干扰影响。相反,就持续时间和质量而言,睡眠不足会增加神经退行性疾病的进程,并加重潜在的临床症状。几项研究表明,睡眠中断是神经病理学的主要贡献者。健康受试者一夜睡眠不足或NREM睡眠中断会增加脑脊液中Aβ1-42和Aβ1-40的水平。在小鼠中,睡眠剥夺导致脑间质液中Aβ肽增加,β淀粉样蛋白与失眠有直接关系。
注射食欲素(与觉醒有关的主要神经肽)导致Aβ升高,而食欲素拮抗剂阿莫沙克降低了Aβ水平。睡眠中断和τ病理学之间也可能存在显著的关系,如在睡眠剥夺的AD小鼠模型中发现的受损记忆、τ代谢和突触完整性所示。睡眠中断和Aβ和τ清除受损之间的可行联系是一个功能失调的淋巴系统(图3)。大脑细胞外空间中溶质的运动是由血管周围星形胶质细胞通过水通道蛋白-4(AQUP 4)通道驱动的水交换和血管腔的变化引起的。水通道蛋白4主要在星形胶质细胞的足部表达。并且水通过水通道蛋白4负责通过间质液的对流在动脉旁和静脉旁空间之间主动驱动的流体交换。据推测,小动脉搏动以及依赖于呼吸的小静脉萎陷是造成这种对流流动的原因。脑脊液和组织液之间的溶质交换主要发生在慢波睡眠期间,此时皮质间隙增加超过60%,为脑脊液和组织液在脑实质中的运动提供了低阻力途径。
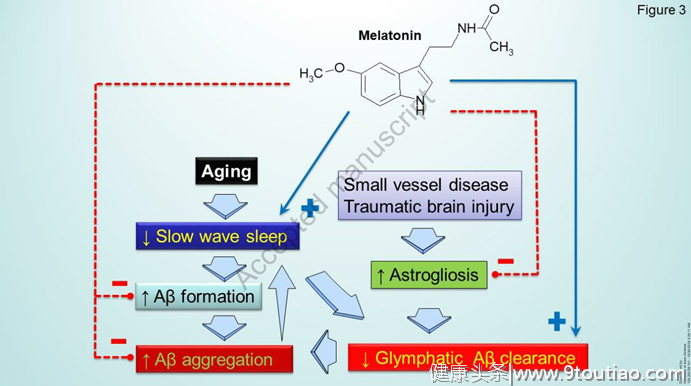
淋巴系统清除Aβ的概念得到了观察结果的支持,即注射放射性标记的Aβ肽的消除在AQP4通道敲除小鼠中显著减少。事实上,水通道蛋白4在血管周围星形胶质细胞脚的位置在AD中被认为是高度改变的,从这个角度来看,AD的发展和进展可能是由于睡眠障碍加重的Aβ清除失败。另一个与AD相关的方面是指从细胞外间隙(ECS)清除载脂蛋白E,因为睡眠剥夺被发现抑制这一过程以及从ECS清除τ蛋白。尽管淋巴假说仍然是一个有争议的问题,正常睡眠对大脑正确清除有毒物质的作用可以被认为是已经确立的(图3)。
AD中的褪黑素
昼夜节律系统的严重破坏发生在AD中,表现为许多身体节律的改变,如体温、血浆糖皮质激素和血浆褪黑激素。这种昼夜节律紊乱的一个新症状是“日落”,这是一种在患有AD以及睡眠和觉醒障碍的患者中观察到的时间生物学现象。时间治疗干预,如暴露在强光下和在选定的昼夜节律阶段定时服用褪黑激素,可以减轻“日落”并改善AD患者的睡眠-觉醒模式。松果体甲氧基吲哚褪黑激素是视交叉上核(SCN)时钟的同步器。
在哺乳动物中,褪黑激素在松果体中以有节奏的方式合成,夜间水平高,白天水平低。褪黑激素通过作用于在SCN神经元中表达的MT1和MT2褪黑激素受体来改变SCN中的昼夜节律,从而在SCN和松果体之间产生相互作用。褪黑激素分泌的昼夜节律已被证明是正常和失明受试者睡眠/觉醒节律的原因(即,在没有光的同步效应的情况下)。褪黑激素显示出从单细胞生物到高等脊椎动物惊人的系统发育保守性,这强烈表明了细胞保护功能。褪黑激素的实验治疗已被证明在衰老和AD动物模型中具有神经保护作用,因为它的给药减少了Aβ和过度磷酸化tau的积累,改善了神经可塑性和神经元存活,防止了学习和记忆障碍,并改善了焦虑和抑郁样行为Pappolla等人的早期工作表明,褪黑激素有效地减少了Aβ的产生和沉积(图3)。
褪黑激素的施用也增加了β淀粉样蛋白的清除率。由于褪黑激素仅抑制Aβ聚集的第一阶段(成核阶段),但一旦形成低聚物或原纤维,就不会恢复原状,因此应考虑将其用于预防AD的治疗应用。关于tau,褪黑激素通过PI3K/Akt/GSK3β信号在小鼠hipoccampus中有效抑制Aβ诱导的tau蛋白过度磷酸化。
炎症和抗炎信号之间的不平衡是导致AD进展的神经退行性过程的标志。引入术语“炎症”是为了强调炎症在衰老中的重要性及其在年龄相关疾病发展中的作用。褪黑激素逆转炎症发生在不同的水平。褪黑激素通过逆转胰岛素信号转导关键步骤的阻断,即降低胰岛素受体底物(IRS)-1的磷酸化,有效抑制胰岛素抵抗(代谢综合征的标志)。褪黑激素抗炎活性的另一个重要方面是其作为免疫缓冲剂的作用,包括促炎和抗炎作用。在衰老和癌症等几种情况下,褪黑激素的抗炎作用占主导地位。至于食欲调节,褪黑激素的可能参与已经研究了多年。褪黑激素调节大鼠、小鼠、仓鼠、猪和几种亚哺乳动物如金鱼、虹鳟鱼和斑马鱼的食物摄入。
在大鼠中,相互矛盾的反应包括减少、增加或不影响食物消耗。据报道,大鼠的脂肪量和体重减少,而褪黑激素增加灰鼠狐猴、叙利亚仓鼠、浣熊和花园睡鼠的脂肪量。在一项评估肥胖大鼠内侧基底下丘脑(MBH)NPY、瘦素受体(瘦素-R)、POMC、催乳素释放肽(PrRP)、胰岛素受体、IRS-1和IRS-2基因表达的研究中,报告说,用2.3毫克/千克褪黑激素治疗10周后,抑制了MBH增加的NPY、瘦素-R、PrRP、胰岛素-R、IRS-1和IRS-2的mRNA水平。这些结果表明,褪黑激素的施用可能能够在下丘脑摄食信号之间建立新的平衡(图1和3)。值得注意的是,褪黑激素降低了强烈食欲素能信号NPY和异常食欲素能信号PrRP的基因表达,以及异常食欲素信号受体(如瘦素和胰岛素)和胰岛素细胞内信号(IRS-1,IRS-2)的基因表达。这种效应是否与临床证明的AD患者褪黑素活性相关值得进一步探讨。
在AD临床阶段进行的褪黑激素试验未能显示出或仅显示出对认知的适度积极影响。基于临床前数据,褪黑激素更有可能阻止Aβ的聚集,而不是逆转疾病临床表现阶段的神经病理学。正常衰老的特征是认知能力下降,包括推理、记忆和语义流利性,这在生命的第五个十年已经可以检测到。有证据表明,与正常衰老(轻度认知障碍)相比,痴呆的临床前阶段认知能力有限。在基于社区的研究中,生活在社区中的老年人样本中有近30%表现出不能用与年龄、教育水平、情绪或健康状况相关的变化来解释的表现缺陷。
这一发现强烈表明早期病变的存在:一种发生在正常衰老和早期AD之间的过渡状态。对已发表的关于在认知衰退早期服用褪黑激素的数据进行分析,结果一致表明,每天晚上在退休前服用褪黑激素,可以改善睡眠质量和疾病这一阶段的认知表现。在轻度认知障碍患者中,褪黑激素的作用是显而易见的,可显著改善睡眠和生活质量,减少认知障碍]。即使在AD的临床前阶段,当患者没有表现出认知障碍时,脑脊液褪黑素水平也会降低,这表明脑脊液褪黑素的减少可能是AD的早期触发因素和标志。虽然还不知道相对褪黑激素缺乏是神经退化的结果还是原因,但显然褪黑激素的缺乏会加重疾病,早期昼夜节律紊乱可能是一个需要考虑的重要缺陷。最近的一项研究观察到轻度认知障碍和AD患者之间褪黑素水平的显著差异,痴呆的神经心理学评估和褪黑素水平之间存在负相关。两项荟萃分析支持褪黑激素疗法对改善痴呆症患者的睡眠有效的观点。此外,据报道,褪黑激素能激动剂ramelteon对治疗谵妄有效,谵妄是一种急性精神混乱状态,可导致重症监护病房老年患者出现许多不良后遗症。
结论
本文讨论的证据支持AD患者的下丘脑显著改变,导致能量稳态障碍和代谢障碍,体重减轻与疾病进展相关。事实上,肥胖、胰岛素抵抗和2型糖尿病是AD发展的重要危险因素。此外,AD患者的睡眠障碍发生率接近70 %,约50 %的AD患者在下午晚些时候至晚上早些时候出现神经精神症状恶化(“日落”)。睡眠/觉醒周期的这种中断通过影响定向和非定向脑脊液流动,影响正常脑血管周围和非血管周围有毒废物的清除。褪黑激素在人类医学应用中结合了两种特性:时间生物和细胞保护。
许多已发表的研究支持褪黑激素对睡眠的重要时间生物学调节作用。在一项包括19项涉及1683名受试者的研究的荟萃分析中,褪黑激素在减少睡眠潜伏期和增加总睡眠时间方面显示出显著的功效。持续时间更长的试验和使用更高剂量的褪黑激素显示了更大的效果。一些共识声明也支持褪黑激素的这一作用。例如,英国精神药理学协会关于失眠、半睡眠和昼夜节律障碍循证治疗的共识声明得出结论,“……褪黑激素应该是55岁以上失眠症患者的首选安眠药”。2007年,欧洲药品管理局(EMA)批准了一种2毫克褪黑激素的缓释形式(Circadin,Neurim,特拉维夫),用于治疗老年人失眠。对于褪黑激素类似物rame lton(Rozerem,Takeda)和tasimelton(Hetlioz,Vanda),EMA和美国美国食品药品监督管理局强调了褪黑激素没有显示出依赖性、孤立性、反弹性失眠或对白天警觉性的负面影响的事实。
关于细胞保护,几乎人体的每一个细胞都含有褪黑激素,其含量远远高于来自松果体的血液循环中的褪黑激素。要改变细胞内褪黑激素水平,需要的剂量要远远高于作为时间生物剂使用的剂量(即在40-100毫克/天的范围内)。鉴于在AD动物模型中的研究,显而易见的是,褪黑激素的几个潜在有用的作用,例如防止τ磷酸化的Aβ形成,需要褪黑激素的剂量在大于100毫克/天的量级,作为等效的人剂量。
如果我们期望褪黑激素能有效改善健康,特别是在老年人中,很可能目前通常施用的低剂量褪黑激素(低于10mg/天)是无益的。已发表的报告表明,褪黑激素是一种安全、低毒性的药物。在健康志愿者的两项褪黑激素剂量递增研究中,评估了高达100毫克口服剂量褪黑激素的耐受性和药代动力学,未检测到任何副作用。然而,褪黑素在长期治疗中的安全性仍有待解决。食物中褪黑激素的富集可以提供一种策略,达到在AD中提供有效细胞保护的量。因此,一个感兴趣的领域是开发含有高水平褪黑激素的功能性食品。褪黑激素在世界上许多国家被广泛用作食物补充剂、食疗产品和药物。欧洲食品安全局(EFSA)已经承认了褪黑激素减少睡眠开始潜伏期的健康声明。因此,富含褪黑激素的食物和生物提取物现在可以被开发成营养补充剂、食疗产品和药物。
本文转载自其他网站,不代表健康界观点和立场。如有内容和图片的著作权异议,请及时联系我们(邮箱:[email protected])