DNA甲基化与癌症之NGS检测篇
什么是甲基化
在哺乳动物细胞中,DNA甲基化的特征是在DNA甲基转移酶(DNMT)的作用下,胞嘧啶嘧啶环(5-甲基胞嘧啶;5mC)第5位碳原子上添加上甲基基团(-CH3),并且甲基的共价加成通常发生在CpG二核苷酸中的胞嘧啶中(见下图[1]),该二核苷酸集中在称为CpG岛中,CpG位点成簇的以预期频率出现。在人类基因组中,约有2800万个CpG位点,约70%的正常体细胞中为甲基化,并且,这些CpG位点的分布并不均匀。大多数CpG岛的长度为500-1000个碱基对(bp),他们通常跨越启动子区域,特别是管家基因。与大部分基因组不同,位于CpG岛内的CpG位点通常在正常体细胞中为未甲基化状态。
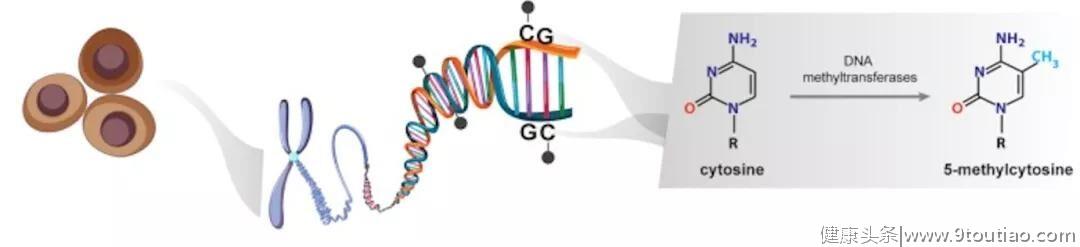
在哺乳动物中,负责5-甲基胞嘧啶(5mC)形成的DNA甲基转移酶包括DNMT1,DNMT3A及DNMT3B。DNMT1酶可以识别半甲基化的DNA,并保证维持现有的甲基化模式;DNMT3A和DNMT3B会在未甲基化的胞嘧啶上添加甲基基团。除了5-甲基胞嘧啶(5mC),5-羟甲基胞嘧啶(5hmc)为已发现胞嘧啶的甲基化中间产物,与5-甲基胞嘧啶(5mC)都为哺乳动物基因组中发现的两种最常见的表观遗传标记,通过双加氧酶家族TET(包括TET1、TET2和TET3)蛋白,将5-甲基胞嘧啶(5mC)氧化为5-羟甲基胞嘧啶(5hmc)[2]。DNA的5-甲基胞嘧啶(5mC)和5-羟甲基胞嘧啶(5hmc)水平,在肿瘤发生发展中发挥了重要的作用。
甲基化与癌症
DNA甲基化对于发育以及维持细胞正常功能至关重要,肿瘤细胞中甲基化特征与正常细胞大有不同,并且,在癌症患者中可以同时检测到低甲基化和高甲基化水平的改变。在肿瘤的发生的初始及进展阶段,表观水平就已经出现了异常,DNA甲基化整体上发生了改变。一般而言,CpG区域的甲基化水平总体下降,可导致基因组的不稳定性,但较少地激活沉默的原癌基因[1]。
癌症的发生、发展伴随着DNA甲基化模式的改变,包括了逆转录元件、着丝粒及原癌基因的DNA低甲基化,以及与基因抑制相关的关键基因调控元件(如远端增强子和启动子转录起始的重叠区域)的甲基化。如下图所示,在整个基因组的所有基因调控元件,正常细胞和癌症细胞之间的DNA甲基化模式存在广泛差异。正常基因组中的大部分CpG位点都携带着5mC,而远端增强子元件及CpG岛区域对甲基转移酶DNMT的活性具有抗性。癌细胞主要表现特征为整体范围的失去甲基化遗传学修饰,反而增强子和启动子区域内出现异常的甲基化位点。这种甲基化分布的改变,导致了肿瘤抑癌基因的表达受抑制,并伴随着原癌基因表达的增加,从而进一步推动了肿瘤的发生、发展。(在下图中,白色圆圈代表未甲基化CpG位点;黑色圆圈代表甲基化CpG位点[1])。
DNA甲基化为常见的肿瘤标记物
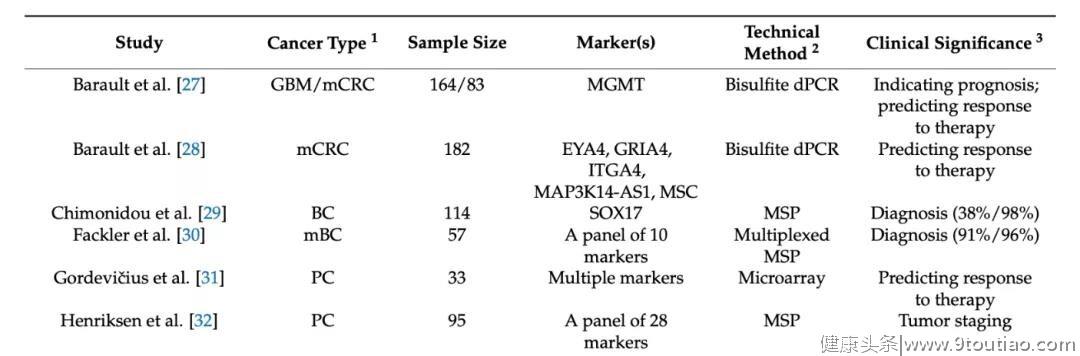
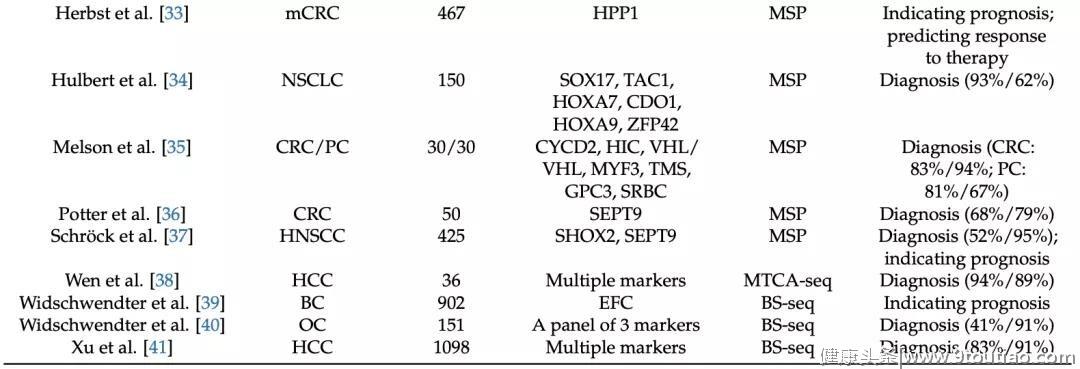
二十多年前,卢煜明教授等人已经证明了利用DNA甲基化作为生物标记物检测孕妇/癌症患者血浆中胎儿/肿瘤源性DNA的可行性[3,4]。研究表明,在妊娠和癌症模型中,cfDNA与其组织来源的基因组DNA之间的DNA甲基化特征高度一致。DNA甲基化在同一个体具有高度的组织特异性,同时,不同个体的相同组织,其甲基化水平可能也具有着一致性[5]。可将这一概念应用于分析血浆样本中的DNA甲基化水平,就可以推测cfDNA的组织起源。与检测遗传水平突变相比,表观遗传修饰在不同癌症种类中具有更高的一致性。在临床实践中,可以从实体瘤或癌症患者的血浆DNA中挖掘出具有临床价值的DNA甲基化生物标记物。Wanxia Gai等人总结了近些年利用此类DNA生物标记物进行癌症检测的研究(见下表) [2]。
甲基化检测应用
提到甲基化在癌症领域的应用,最值得分享的便是长期专注于癌症早诊早筛的医疗公司GRAIL及将MRD用于早期结直肠癌患者的检测和复发监测的Guardant Health(GH)。
去年六月,GRAIL在欧洲肿瘤学会(ESMO)旗下期刊《肿瘤学年鉴》(Annals of Oncology)发表了CCGA(Circulating Cell-free Genome Atlas,CCGA)的三个子项目实验结果。在CCGA1的原理发现阶段,使用训练集样本1785例及验证集样本1015例,同时对三种不同的高通量测序(NGS)方案进行评估,包括了靶向测序、全基因组测序(拷贝数变异,CNV)及全基因组亚硫酸氢盐测序 (WGBS)。验证结果证明, 相比于DNA突变水平检测,WGBS技术路线更适合应用于癌症早筛的研究领域[6]。GRAIL也同时公布了其结合机器学习算法的cfDNA的甲基化测序分析技术可以同时检测五十多种癌症类型,其检测的特异性>99%,并且对肿瘤信号组织来源的预测准确性>90%[6]。由于 CCGA 是一项病例对照研究,可能无法真实的反映该血液检测在普通人群筛查情况下的表现。目前,研究团队仍在持续进行 PATHFINDER 实验,通过纳入更多的健康人群,以评估检测方案在临床护理环境中的实施及性能。
甲基化应用的另一个方向为MRD(Minimal Residual Disease),即微小残留病灶(是指癌症治疗后残留在体内的少量癌细胞(对治疗无反应或耐药的癌细胞))。今年四月,《临床肿瘤研究》(Clinical Cancer Research)在线发表了名为《Minimal Residual Disease Detection using a Plasma-only Circulating Tumor DNA Assay in Patients with Colorectal Cancer》文章,Guardant Reveal首次公开了仅使用血浆ctDNA MRD的检测数据,证明MRD检测灵敏度达到91%,特异性达100%。Guardant Reveal的MRD技术路线是Tumor-agnostic策略,又称为Tumor-uninformed,即仅依赖于血浆ctDNA进行MRD检测(无需组织活检),检测方案为整合ctDNA突变(LOD为0.01%ctDNA)及ctDNA甲基化水平检测。该研究表明,通过整合表观基因组标记,如DNA甲基化分析,比单独基因组水平突变检测灵敏度更高[7]。
甲基化检测之NGS方法及流程
随着高通量测序技术的发展,NGS(Next-generation sequencing)也成为一种可快速获得任何物种DNA甲基化图谱的检测方法。除了选择全因组范围内的甲基化水平检测外,还可以进行DNA富集后再测序,如MeDIP或MBD Cap(也称为MeDIP-seq或MBD-Cap-seq)。甲基化DNA片段被特定的抗体或蛋白质捕获 ,经过纯化、扩增等步骤后测序。但该种检测方案不能准确的识别单个位点的甲基化水平,常用于评估特定区域的甲基化水平。与之类似的为基于限制内切酶的前处理方案,称之为MSRE(MSRE-seq),使用甲基化位点敏感的限制性内切酶(BstUI、HpaII、NotI和SmaI),针对非甲基化限制位点进行切割,同样经过纯化、扩增等步骤后测序。由于以上两种方法都具有较低的检测分辨率和基因组覆盖率,也就限制了在肿瘤领域,特别是癌症早筛、早诊的临床应用[8]。
重亚硫酸氢盐处理一直是绘制DNA甲基化图谱的金标准,由来自澳大利亚的Kanematsu等科学家开发的技术方案[10,11],DNA经重亚硫酸氢盐处理后,其胞嘧啶残基转化为尿嘧啶残基,5-甲基胞嘧啶(5mC)则保持不变,各检测方案对比见下表[9]:

如果想针对大量样本的特定区域进行甲基化检测时,WGBS无疑成为一个成本相对较高的技术方案。幸运地是,杂交捕获技术可以在一次反应中检测成千至上百万个目标基因组序列碱基,为此提供了一种性价比极高的解决方案,在成本允许的情况下可以实现更高的测序深度和更大规模的研究。
常规甲基化文库制备方法为下图中流程B所示,DNA在连接反应步骤中加入测序接头,转化成可以上机测序的文库后进行重盐硫酸盐处理,也称作PreBS(Pre-Bisulfite)方法。该方案通常需至少微克量级别DNA,其主要原因是亚硫酸氢处理会破坏DNA双链结构,导致测序时损伤的文库无法进行簇生成。文库序列信息或独特分子的大量丢失,限制了供人类疾病研究的样品类型,如游离核酸。即使低起始量DNA样本最终转化成可以上机的测序文库,但由于重盐硫酸盐处理而导致极低的文库分子复杂度,影响了最终的测序质量,如产生较高的dup率。
为了解决上述遇到的难题,重亚硫酸氢盐处理后进行建库的技术方案逐渐成为主流的检测路线,即PostBS (Post-Bisulfite),PBAT(Post Bisulfite Adapter Tagging)也属于该方法之一(流程C)。PBAT是以重亚硫酸氢盐转化后的单链DNA为初始模板,通过两轮随机引物延伸,从而连接两端测序接头。由于随机引物的使用,使得:
1
重亚硫酸盐处理后所产生的损伤DNA模版, 其中有一定的比例无法结合引物,从而降低了测序文库的分子复杂度;
2
随机扩增有着模版偏好性。
值得注意的是,PBAT方案的改良版本也一直用于单细胞甲基化分析方案中。
Swift Biosciences(IDT埃德特公司收购)创立了更加优化的建库PostBS 流程,与PBAT相同,都以重亚硫酸氢盐转化后的单链DNA为初始模板;不同的是,IDT xGen™ Methyl-Seq Library Prep(原来名称Accel-NGS Methyl-Seq Library Kit,Adaptase技术)对重亚硫酸氢盐转化处理后的单链及损伤DNA进行最大程度地利用和转化,从而提供更完整、偏好性更低的甲基化文库(流程A)。

IDT xGen™ Methyl-Seq Library Prep基于Adaptase®专利技术,该技术是一种高效的、不依赖于模板的单链DNA接头连接方法。该建库试剂盒提高了文库转变效率,允许将重亚硫酸氢盐转化处理后的DNA样品连接测序接头,对样本组成实现精准呈现。通过高效率的接头连接方式,对重亚硫酸氢盐转化处理后的单链及损伤 DNA 进行文库构建,相比构建文库后再做亚硫酸氢盐转化处理的方法,文库产量可提升100倍;除此之外,使用IDT特有的、无偏好性的接头连接方式,经全基因组亚硫酸氢盐测序 (WGBS)验证,xGen™ Methyl-Seq Library Prep甲基化建库试剂盒可提供更完整、更偏好性更低的NGS文库。
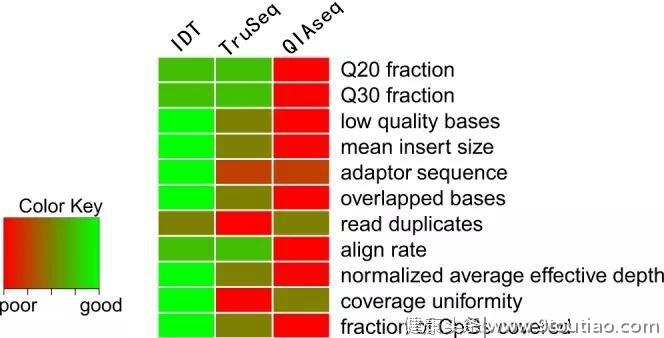
来自新加坡南洋理工大学Li Zhou等人在Illumina NovaSeq和HiSeqX测序平台上,系统的比较了三种PostBS文库制备试剂盒性能,包括xGen™ Methyl-Seq Library Prep, Illumina TruSeq DNA Methylation kit (PBAT)和QIAGEN QIAseq Methyl Library kit (PBAT)[12]。实验设计如下表所示,样本1-4及样本5-8分别为全血和白细胞亚群(样本5和8:CD4+T细胞;样本6和7:中性粒细胞)
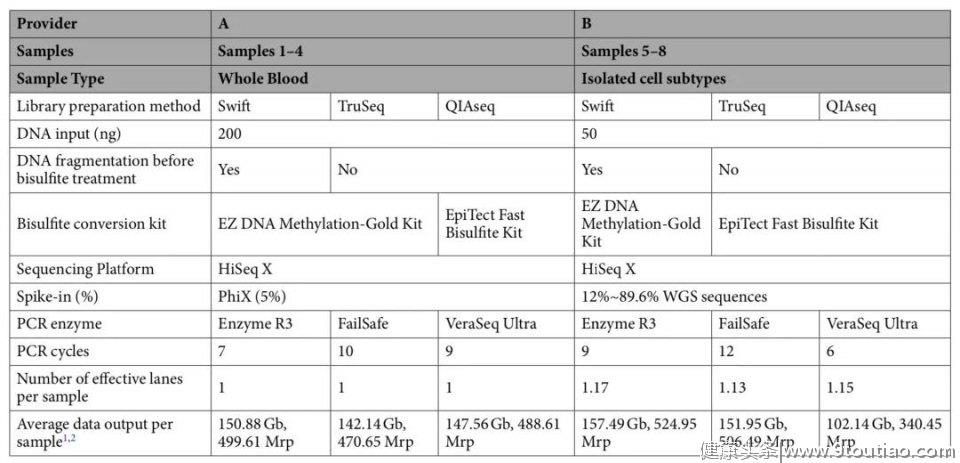
该研究团评估了包括测序质量(Q20, Q30, 低质量碱基去除比例等)、文库质量(平均插入片段大小、重叠区域比例、dup率等)、覆盖度(平均有效测序深度、覆盖度均一性、CpG位点覆盖情况)等测序指标,下方热点图综合了三种建库制备方法的总体性能比较结果,其中“绿色”和“红色”分别表示性能表现好及差的技术方法。如某一特定指标两两比较时,在统计上没有显著差异,那么该两种方法将被赋予平均表现的相同等级,例如,在评估Q20测序指标时,IDT xGen™ Methyl-Seq Library Prep和TruSeq都为最佳表现的试剂盒,且等级分数相同(2.5=(2+3)/2),因为两者之间没有统计学上的显著差异。
总结
甲基化测序是研究不同生命过程中基因调控的一个重要工具,例如细胞分化和疾病进展,并且越来越多的应用到包括肿瘤早筛、分诊、治疗选择、微小残留监测、复发检测等临床领域中。包括游离核酸在内的体液样本,含有来自于肿瘤的特异的DNA甲基化信号,作为生物标志物样本检测来源,无疑是一个绝佳的选择。
重亚硫酸氢盐处理一直是绘制DNA甲基化图谱的金标准,由于其转化效率的稳定性,保证了后续甲基化检测结果的准确性。在针对特定的目标区域进行大样本量的检测时 ,WGBS就成为一个成本相对较高的技术方案。杂交捕获技术,搭配PostBS单链建库流程,可对感兴趣的目标区域进行精准研究。
参考资料:
1. Skvortsova K, et al. The DNA methylation landscape in cancer. Essays Biochem. 2019 Dec 20;63(6):797-811.
2. Giorgia Gurioli. Epigenetic Characterization of Cell-Free DNA. Cell-free DNA as Diagnostic Markers pp 129-135.
3. Poon, L.L, et al. Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin. Chem. 2002, 48, 35–41.
4. Wong, I.H.; et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999, 59, 71–73.
5. Kundaje, A.,et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330.
6. Liu MC, Oxnard GR, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol. 2020 Jun;31(6):745-759.
7. Aparna R Parikh, et al. Minimal Residual Disease Detection using a Plasma-only Circulating Tumor DNA Assay in Patients with Colorectal Cancer. Ann Oncol. 2020 Sep;31(9):1266-1267.
8. Locke WJ, et al. DNA Methylation Cancer Biomarkers: Translation to the Clinic.Front Genet. 2019 Nov 14;10:1150.
9. Vivek Kumar, et al. (2019) Techniques/Tools to Study Epigenetic Biomarkers in Human Cancer Detection Biomedical Engineering and its Applications in Healthcare pp 327-351
10. Frommer, M., et al. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U. S. A. 89 (5), 1827–1831.
11. Clark, S. J., Harrison, J., Paul, C. L., and Frommer, M. (1994). High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 22 (15), 2990–2997
12. Li Zhou,et al. (2019) Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing. Sci Rep. Jul 17;9.