非小细胞肺癌,ALK突变抑制剂,哪个更好?
ALK融合是晚期非小细胞肺癌(NSCLC)的重要治疗靶点,有“钻石突变”之称。(ALK突变的正在进行的新药临床试验可点文末的【了解更多】)自FDA批准一代ALK-TKI克唑替尼以来,二代ALK-TKI阿来替尼、塞瑞替尼、布格替尼、恩沙替尼以及三代ALK-TKI劳拉替尼陆续丰富序贯用药格局,ALK融合阳性的患者中位总生存期(OS)大大延长,逐步迈入“慢病化”管理时代。新的问题随之而来,百花齐放的ALK抑制剂,风起云涌的一线战场,谁将称霸?
彰明往事而察知来事,本文将带您回顾ALK如何成为癌症治疗的重要靶标,梳理三代ALK-TKI抑制剂的药物发现与历史进程,并基于医药魔方NextPharma、NextMed数据库,快速检索、简要呈现相关药物的循证数据和指南推荐,实现ALK药物直观的对比分析。
追本溯源篇
ALK (Anaplastic lymphoma kinase),即间变性淋巴瘤激酶,属于胰岛素受体(IR)蛋白-酪氨酸激酶超家族的成员,最初于1994年在间变性大细胞淋巴瘤的一种亚型中被发现,由此而得名,该基因融合了整个核磷蛋白(NPM)基因。人ALK基因位于2p23染色体片段上,成熟的ALK蛋白包含胞外配体结合结构域、跨膜结构域和细胞内酪氨酸激酶结构域,在配体诱导作用下形成同源二聚体被激活,具有调节细胞增殖、分化并抑制凋亡等作用。[1]
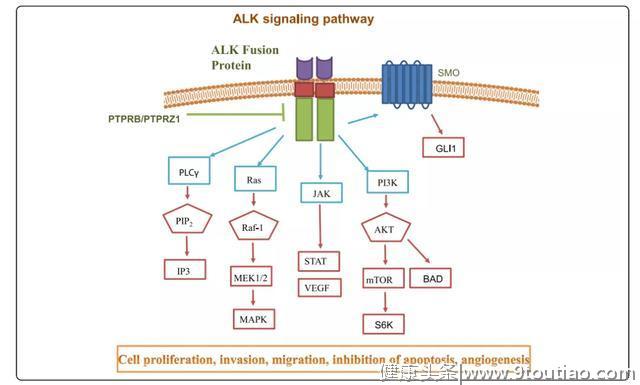
ik2007年,Soda及其同事首次发现NSCLC患者的ALK重排现象[2],约3%~7%的 NSCLC 患者存在ALK重排[3],并且不与EGFR突变同时存在。目前已经发现27种ALK 重排变异体,最常见的是棘皮动物微管相关蛋白4(EML4) -ALK融合基因,它是 EML4的13号外显子上断裂产生的3.6kb大小的片段插入并连接上ALK的20号外显子的一个297bp大小的片段,共同构成了融合基因 EML4-ALK,这种融合基因可导致酪氨酸酶的激活,促进肿瘤细胞的生长分化,也使其成为癌症治疗的重要靶标。EML4-ALK 融合基因至少有11种亚型,其中最主要的亚型是EML4-ALK1(33%)[4]。
虽然ALK基因融合最早在NSCLC患者中发现,并且NSCLC是ALK抑制剂目前最重要和成功的瘤种,但随着研究进展,发现基因融合只是影响ALK基因遗传改变的一部分,而且ALK改变的现象并不单纯出现在NSCLC中。在神经母细胞瘤、胶质母细胞瘤、横纹肌肉瘤、卵巢癌、乳腺癌、无炎性成肌成纤维细胞瘤(IMT)、鳞状细胞癌以及其他多种癌症中,均发现ALK过表达和点突变引起的致癌进展。
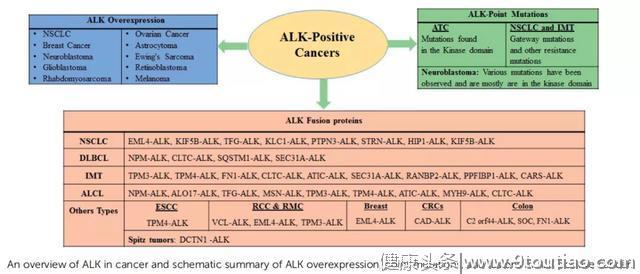
药物发现史
药物的发展历程就是如此,明确致癌驱动因子之后,临床开发便会提上日程。ALK药物至今已经发展到第三代,每一个药物的治疗推荐也是一步步走向了一线,ALK阳性NSCLC一线治疗即将呈现出“三代同堂”的盛况,更重要的是药物的上市已为疾病治疗和预后带来了巨大的改善。
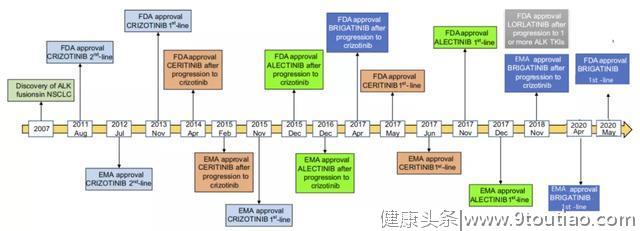
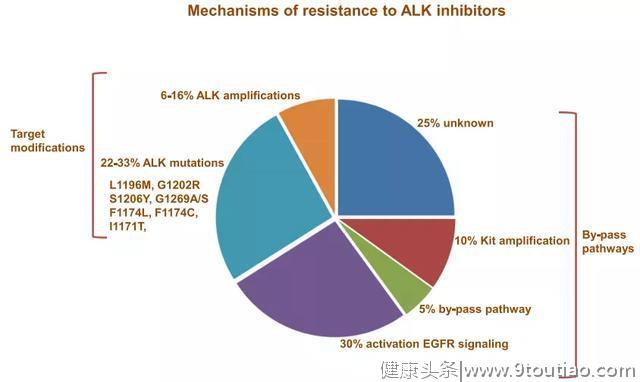
当然,每一代药物的发展也是为了更好的解决抗药性的突变。对ALK抑制剂的抗性包括由ALK激酶结构域中的获得性继发突变(F1174 L,F1174C,L1196 M,I1171T,G1202R,S1206Y,G1269S和G1269A)或ALK基因扩增和其他的激活途径,如EGFR或胰岛素样生长因子途径以及肿瘤组织学类型转化等介导。目前,已获批的ALK药物更多是解决了获得性继发突变。
2011年,也就是Soda等人发现ALK重排作为NSCLC 潜在的致癌驱动因子后仅4年,克唑替尼就被FDA批准用于治疗ALK阳性的晚期NSCLC。作为辉瑞开发的一种口服小分子ATP竞争性ALK抑制剂,克唑替尼最初用作MET-TKI,在发现 ALK重排在NSCLC中的作用后迅速转向ALK。
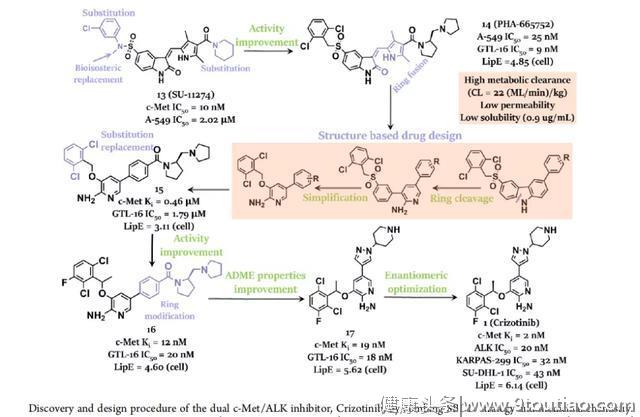
最初,克唑替尼是由辉瑞公司采用药物综合发现策略针对c-Met激酶所设计的。如上图所示,辉瑞公司的研究人员首先确定了一种3位取代的吲哚啉-2-酮衍生物SU-11274,然后将其优化为有效的c-Met抑制剂PHA-665752(舒尼替尼类似物)。由于其理化性质较差,如低溶解性、较差的膜透过性和代谢迅速等,通过基于结构的药物设计(SBDD),利用吲哚酮衍生物PHA-665752与c-Met激酶结构域结合的共晶体结构中揭示的新型ATP位点环镜进行设计,将核心骨架重新设计为新型的5-芳基-3-苄氧基-2-氨基吡啶核,这样的设计使3-苄氧基与母体化合物的2,6-二氯苯基位于同一口袋,但配体效率更高,致使新设计分子活性低。因此,针对活性和ADMET性质开展了进一步的优化,并最终获得了克唑替尼。
克唑替尼在临床试验中表现出很好的效果,约60%的患者可以得到缓解,中位无进展生存期(PFS)为8~10个月。但是,约30%的ALK阳性NSCLC患者对克唑替尼先天耐药,剩下70%在治疗约1年后都表现出对克唑替尼不同程度的耐药,这种后天获得性耐药主要包括药理学耐药及生物学耐药。
药理学耐药的主要原因是克唑替尼几乎不能透过血脑屏障,生物学耐药主要是药物作用靶点以及 ALK信号通路的改变,药物作用靶点改变包含ALK 激酶区突变(28%)以及ALK融合基因拷贝数扩增(9%),约30%~45%患者经克唑替尼治疗后会发生ALK激酶区突变以及ALK融合基因拷贝数扩增,最常见的获得性突变是L1196M、G1269A/S、C1156Y,G1202以及1151Tins、L1152R、I1171T/N/S、F1174 V等,所有这些都在呼吁着下一代ALK药物的出现。
塞瑞替尼是诺华开发的第一个二代ALK抑制剂上市药物,显示了对克唑替尼耐药突变L1196M、G1269A、C1156Y、S1206Y、I1171T 、F1245C等的显著活性。
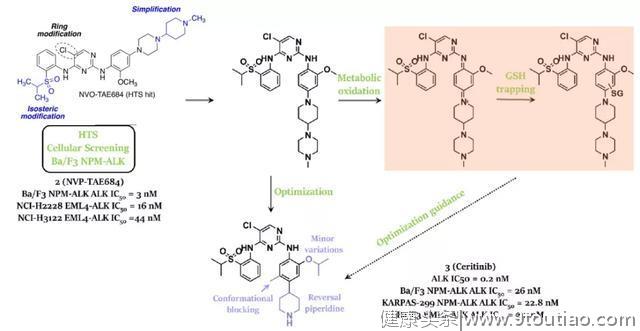
2013年,诺华制药公司的Marsilje及其同事报告了发现带有2,4-二氨基嘧啶核心骨架的化合物3,该骨架来源于对高通量筛选(HTS)目标化合物2(NVP-TAE684)的结构优化。TAE684对带有NPM-ALK的Ba/F3和两个带有EML4-ALK的NSCLC细胞系(NCI-H2228和NCI-H3122)表现出强大的效价。但是,如上图所示,化合物2由于其潜在的氧化代谢毒性而未能进入临床研究,根据结构-活性关系(SAR)的分析进一步证实,活性代谢产物(1,4-二氨基醌结构)的产生主要归因于中间苯胺部分上氮原子连接的可溶性基团。因此,进行了进一步的修饰,以提高激酶的选择性并阻止反应性代谢产物的形成,最终产生化合物3(塞瑞替尼)。
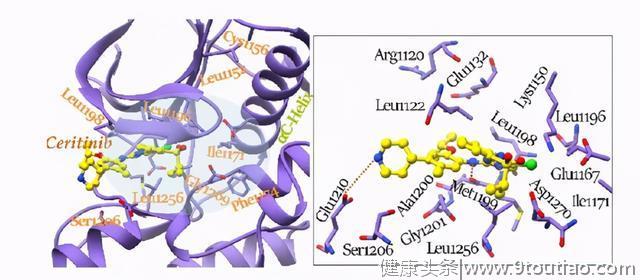
化合物3与铰链区残基Met1199形成两个H键,并且反向的哌啶环与Glu1210以盐桥作用。异丙氧基可以与Arg1120、Glu1132等铰链区形成良好的相互作用,由此,化合物3的选择性和效能均得到提升。如预期的那样,化合物3保留了对ALK的高效能,IC50值为0.2 nM并显示出对带有NPM-ALK融合基因的Karpass-299和Ba/F3细胞系有效抗增殖活性,IC50值分别为22.8和26.0 nM。此外,化合物3可以在H2228大鼠异种移植模型中诱导肿瘤完全消退,并且部分地在Karpass-299异种移植模型中抑制肿瘤的进展。
三项多中心I/II期试验(ASCEND-1,-2和-3)和两项随机III期临床试验(ASCEND-4和-5)均有力证实塞瑞替尼对克唑替尼未治疗和耐药的具有ALK重排的NSCLC患者的治疗效果。然而,塞瑞替尼对G1202R和F1174V/C无抑制性,体外实验也证实1151Tins 和 L1152R 介导了塞瑞替尼的耐药。
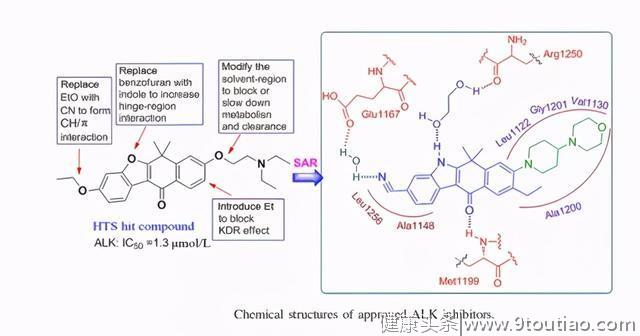
阿来替尼是独特的第二代ALK抑制剂,最初日本的Chugai公司(罗氏子公司)通过高通量筛选发现了一类带有苯并[B]咔唑酮母核结构骨架先导化合物,为改善结合力提高活性用吲哚部分取代苯并呋喃片段,进一步通过优化代谢稳定性、溶剂相互作用以及调节ATP结合位点,以改善激酶效力、选择性和药代动力学特性,从而发现了化合物阿来替尼。
临床前研究证实,阿来替尼的 ALK抑制活性高于克唑替尼,后续体内外试验也进一步确证了阿来替尼对已知大多数ALK耐药类型有效(除 G1202R)。更重要的是,与克唑替尼和塞瑞替尼比较,阿来替尼不是P-糖蛋白的底物,能够很好的通过血脑屏障。临床前试验中,阿来替尼在大脑与血浆中的浓度比值约为0.63~0.94,塞瑞替尼为0.15,而克唑替尼为0.0026。
临床试验中,87例ALK 阳性经克唑替尼治疗失败的NSCLC 进展期患者给予阿来替尼 600 mg po bid,48%的患者获得持久的客观有效率,中位反应时间(DOR)为13.5月,75%的脑转移患者获得客观缓解(PR+CR),颅内中位反应时间(IDOR)为11月。
2020年ASCO年会上,阿来替尼对比克唑替尼一线治疗ALK阳性晚期NSCLC疗效和安全性Ⅲ期ALEX研究数据更新,研究成果近期在《肿瘤学年鉴》(Annals of Oncology)正式发表。阿来替尼组研究者评估(INV)的无进展生存期(PFS)最终定格在34.8个月,显著优于克唑替尼组的10.9个月(HR=0.43, P<0.0001)。亚组分析结果显示,不论基线是否合并中枢神经系统转移,阿来替尼均显示出PFS的获益。尽管最终的OS数据尚不成熟,但在第5年时,阿来替尼组显示出有临床意义的OS改善(62.5% vs 45.5%)。[5]
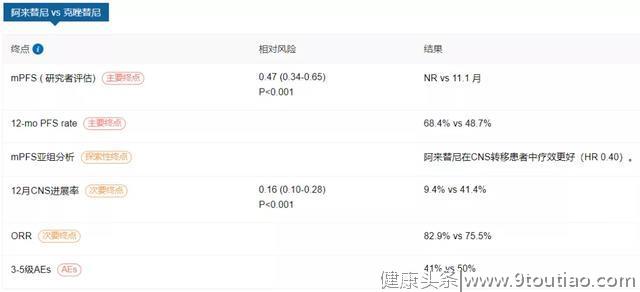
布格替尼也是一种新型 ALK抑制药物,表现出对大多数已知的ALK临床抗药性突变的抑制作用,并且对G1202R、G1202缺失显示中等抑制,优于克唑替尼、塞瑞替尼和阿来替尼。

2016年,Huang和Ariad Pharmaceuticals的同事报告了一系列基于2-氨基嘧啶并含二甲基氧化膦(DMPO)的ALK抑制剂,布格替尼便是其中的化合物之一,相比未取代的类似物具有更高的活性和选择性。Ariad的ALK抑制剂开发采用了几条并行的策略,包括基于克唑替尼的结构修饰和构建2-氨基嘧啶化合物库。为了发现高效的ALK抑制剂,首先探索了将DMPO引入苯胺的C4’位置并保留C2’为甲氧基,保留甲氧基的目的旨在通过与邻近铰链的疏水小口袋相互作用,令人欣慰的是DMPO的引入确实提高了活性,但选择性不佳。通过对结构-活性关系(SAR)的分析,同时考虑对化合物溶解性的影响,进一步构建包含不同取代的化合物库,以提高活性和选择性。最终发现了化合物5(布格替尼),化合物5表现了对InsR和IGF1R良好的选择性。同时,化合物5的高效价得以良好保留,甲氧基与Leu1198、C5-氯原子与Leu1196以及二甲基氧化膦(DMPO)与DFG基序部分之间的相互作用增强。此外,独特的DMPO充当H键受体,有效促进分子内相互作用并稳定其U形构象。
2016年,一项II期、多中心、随机研究(ALTA)表明,对于克唑替尼耐药的晚期或转移性ALK阳性NSCLC患者,布格替尼的疾病控制率(DCR)达到86%,而且产生明显的全身(ORR:54%)和颅内(ORR:67%)效应,以及持久的PFS(12.9个月)。
近期,ALTA-1L研究第二次期中分析结果在《临床肿瘤学杂志》(JCO)发表,中位随访时间24.9个月时,独立评审委员会盲法评估的PFS在布格替尼组和克唑替尼组分别为24.0个月和11.0个月(HR=0.49,P<0.0001)。研究者评估的中位PFS分别为29.4个月和9.2个月(HR=0.43,P<0.0001)。[6]
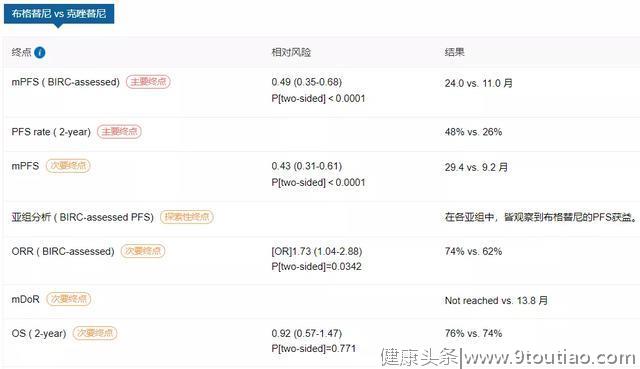
11月11日,医药魔方Plus预警显示贝达药业盐酸恩沙替尼(商品名:贝美纳)的上市申请进入“在审批”阶段,意味着该药有望在不久后正式获批。恩沙替尼是贝达药业自主研发的一种新型强效、高选择性的新一代ALK抑制剂,此次申报的适应症为:用于接受过克唑替尼治疗后进展的或者对克唑替尼不耐受的ALK阳性的局部晚期或转移性NSCLC患者提供新的治疗。
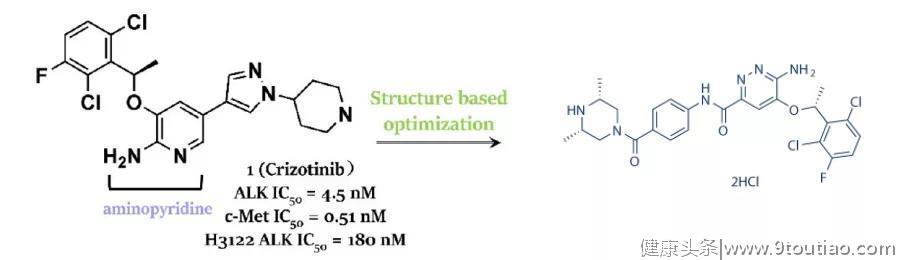
根据其结构,恩沙替尼的来源也是基于克唑替尼,母核结构上体现的设计手段也是药物研发惯用的技术,而对于侧链的修饰恐怕也是基于效价、ADMET方面的考虑。恩沙替尼的一线临床研究也在进行中,今年世界肺癌会议(WCLC)上发表的III期eXalt3试验结果显示,与克唑替尼相比,恩沙替尼的中位PFS明显更长(25.8 vs. 12.7个月;P =0.0003)。在改良的ITT人群中,恩沙替尼的中位PFS尚未达到,而克唑替尼则为12.7个月。在改良的ITT人群中,恩沙替尼和克唑替尼的ORR分别为75%和67%;在有脑转移的患者中,恩沙替尼的颅内ORR为54%,克唑替尼为19%。[7]
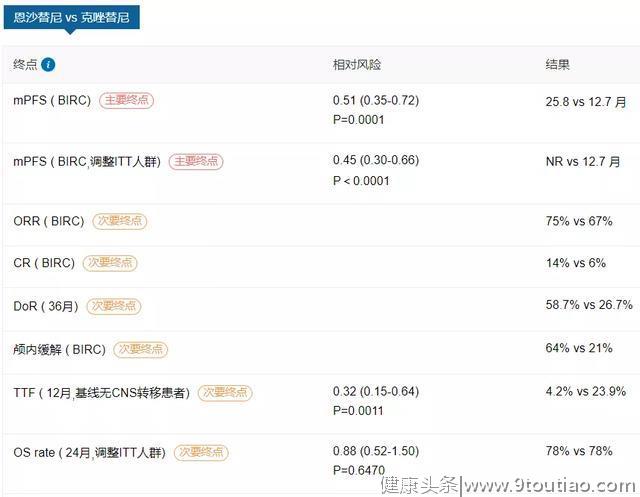
已经获得批准的ALK药物都是具有ATP竞争性的抑制剂,而且分子中均含有延伸至溶剂区域的骨架片段,这些化合物不可避免地易受溶剂前沿突变(SFM)的影响,例如ALKG1202R。
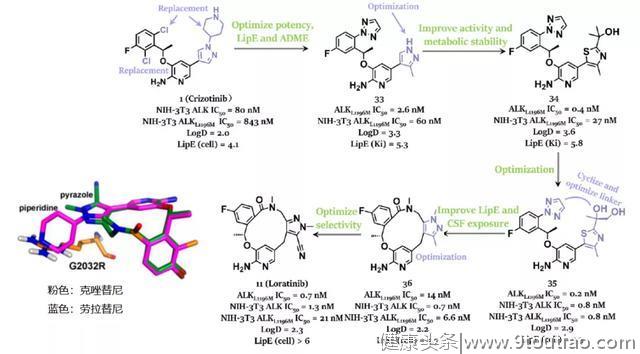
幸运的是,2014年,Johnson等人报道了使用结构引导的方法发现了一种有效ALK抑制剂,该化合物通过酰胺基连接构成13元大环化合物11(劳拉替尼)。在他们的工作中,通过比较apo ALK和ALK克唑替尼复合的晶体结构来设计化合物11,将大环化合物11中的刚性大环以预定的生物活性结合姿势精确锚定在腺嘌呤结合位点,以避免熵损失。此外,化合物11可与Glu1197和Met1199形成两个稳定的氢键,并通过范德华相互作用与DGF-Asp1270形成良好的接触。大环化合物11的最终成功产生归功于提高效力、选择性、CNS ADME和亲脂性效率的一系列努力。
劳拉替尼是一种有效的双重ALK/ROS1抑制剂,对ROS1、ALK和L1196M突变的ALK的Ki值分别<0.02、<0.07和0.7 nM,并且对表达ALKG1202R的Ba/F3细胞显示出良好的抑制活性。值得注意的是,化合物11可以有效地穿过BBB并渗入中枢神经系统(CNS)。根据辉瑞公司发起的I / II期临床试验,劳拉替尼在ALK或ROS1阳性的NSCLC患者中显示出有希望的全身和颅内活性。I/II期试验的结果促进了劳拉替尼在日本于2018年9月首次获批用于耐药性ALK融合阳性晚期和/或复发性NSCLC。
日前ESMO大会期间,劳拉替尼对比克唑替尼一线治疗晚期ALK阳性NSCLC的III期CROWN皇冠研究最终揭晓,根据BIRC评估,劳拉替尼相比克唑替尼带来了显著的PFS获益(未达到 vs 9.3个月),降低疾病进展或死亡风险高达72%,昂首挺进一线。次要研究终点方面,两组ORR分别为76%和58%,值得一提的是,劳拉替尼组的颅内客观缓解率高达82%,完胜克唑替尼组的23%。[8]
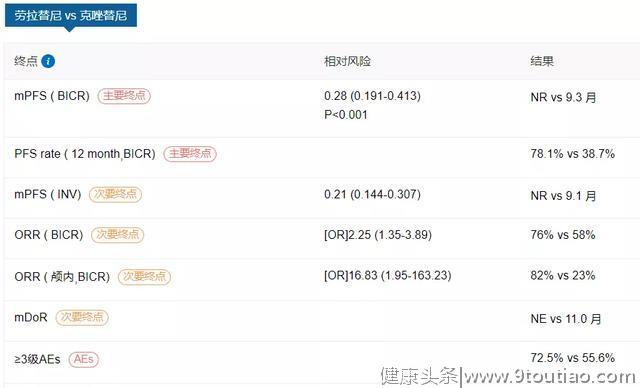
回顾三代ALK-TKI药物的发展,每一种药物均为患者提供了良好的治疗获益;与此同时,不同药物之间也呈现出有差异的耐药机制。
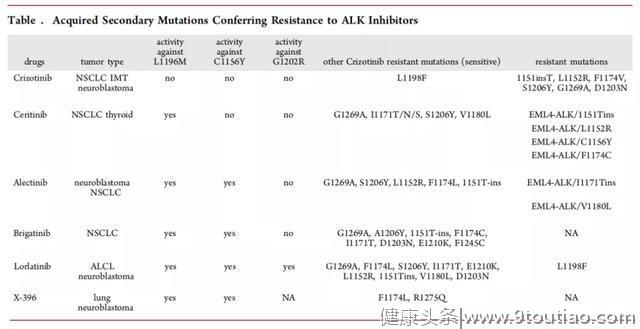
临床应用纪
药物发展的迭代史,也是推动疾病治疗的进步史。虽然国内仅上市三款ALK药物(克唑替尼、塞瑞替尼、阿来替尼),临床用药的选择还不是很多,但已上市的五款ALK药物在国内获批或者说一线用药资格的批准,也许仅是时间上的问题。加之,可以预见的恩沙替尼国内率先上市,以及可能的多款国产ALK药物后续上市,临床医生的选择将会有很大的余地。选择多了,随之而来的问题也就来了,如何选、选哪个、怎么用或许将是面临的新抉择。
临床用药的依据是药物对疾病的治疗功效,患者概况、有效性、耐受性、药物可及性(经济)等因素兼而顾之。

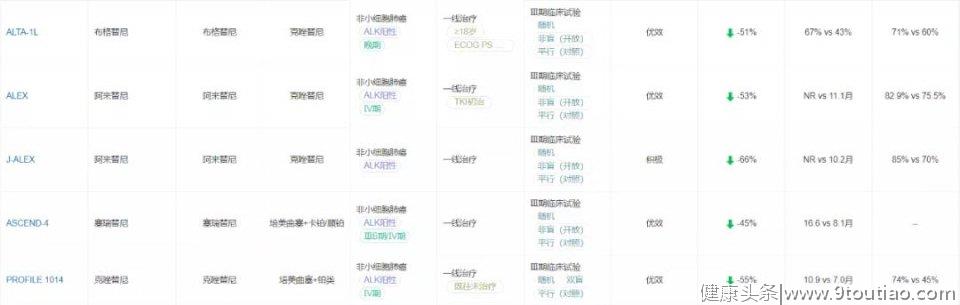
目前数据显示,克唑替尼治疗具有脑转移NSCLC患者的效果较差,这与其血脑屏障通透率较低相对应;而劳拉替尼可有效对抗各类ALK继发的耐药基因突变,并有较强的CNS渗透性。

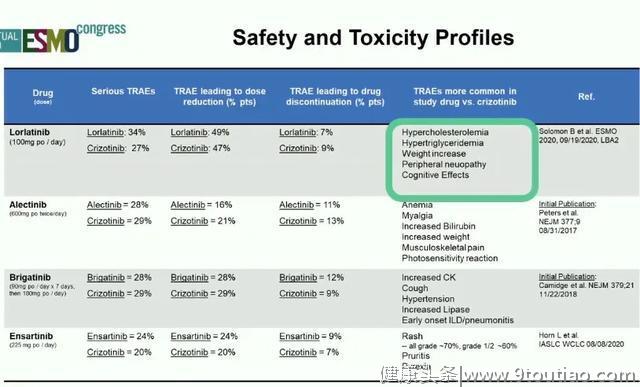
二、三代ALK-TKI之间尚缺乏头对头对比,药物选择时需“因地制宜”,考虑包括治疗持续时间、CNS活性范围、毒性图谱和长期不良事件在内的多种生物学因素。显而易见,未来ALK这个舞台还会精彩纷呈,竞争还会非常激烈。
利用NextMed数据库临床用药对比模块,可将ALK-TKI指南推荐差异及证据来源直观对比如下:
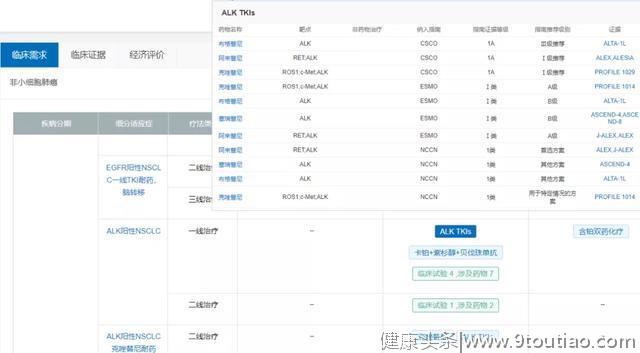
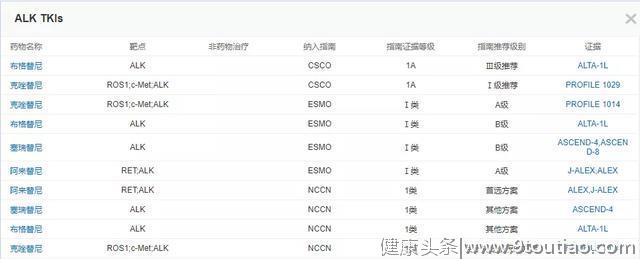
2020年最新版CSCO非小细胞肺癌诊疗指南中,阿来替尼和克唑替尼均作为Ⅰ级推荐,同时阿来替尼备注为优先推荐。含铂双药化疗或含铂双药化疗+贝伐珠单抗(非鳞癌)作为Ⅱ级推荐,布格替尼则作为Ⅲ级推荐。
最新版的NCCN指南中,阿来替尼被作为ALK阳性晚期NSCLC一线治疗的优先推荐,布格替尼和塞瑞替尼作为其他推荐,某些情况下推荐使用克唑替尼。
2019年ESMO转移性非小细胞肺癌临床实践指南中,阿来替尼和克唑替尼均被作为ALK阳性NSCLC一线治疗的A级推荐,同时阿来替尼备注为优先选择;塞瑞替尼和布格替尼则作为B级推荐。
目前,关于ALK 阳性晚期NSCLC患者的真实世界治疗情况和预后数据仍知之甚少,2020年7月发表在《肿瘤学家》(Theoncologist)上的一篇回顾性研究纳入581例接受一线ALK-TKI治疗的晚期NSCLC患者数据,并使用Kaplan-Meier方法计算真实世界无进展生存期(rwPFS)和至停药时间。[9]
该研究中,581例患者接受一线ALK-TKI治疗(27.5%在开始治疗时或之前合并脑转移),254例患者在克唑替尼治疗后接受二线ALK-TKI治疗(45.7%在开始二线ALK-TKI治疗时或之前合并脑转移)。
分析显示,克唑替尼是最常使用的一线ALK-TKI治疗。对于克唑替尼治疗后的二线ALK-TKI,接受塞瑞替尼、阿来替尼、克唑替尼和布格替尼治疗的比例分别为49.6%、41.7%、5.9%和2.8%。一线和二线ALK-TKI治疗的中位rwPFS分别为7.47和7.30个月,脑转移患者的中位rwPFS甚至更短,提示这部分人群需要更有效的治疗。
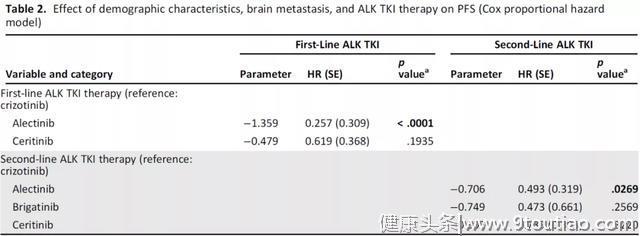
不同ALK-TKI治疗对PFS的影响(COX比例风险模型)
来自美国范德堡大学医学中心的Christine M. Lovly博士在点评CROWN研究时指出,ALK基因检测对于筛选适宜的治疗人群至关重要,研究者正在更广泛的范围开展更多ALK阳性NSCLC临床试验,包括一线联合治疗(如联合免疫检查点抑制剂、VEGFR抑制剂、其他罕见靶点治疗、化疗等)、二线及以上联合治疗、通过循环肿瘤DNA(ctDNA)监测ALK-TKI疗效、将共突变(如TP53)纳入考虑等。
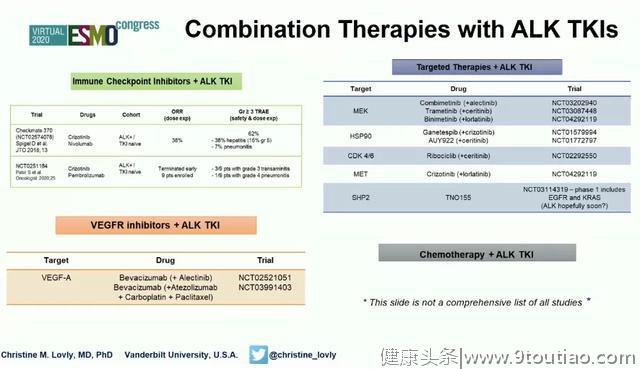
除ALK-TKI外,潜在的ALK靶向治疗包括变构抑制剂、蛋白降解靶向嵌合体技术(PROTACS)和癌症疫苗;ALK-TKI是否可以前移至辅助治疗值得探讨,长期治疗的总生存获益尚待观察;此外,如何将ALK阳性NSCLC的治疗突破扩展至其他伴有ALK重排的肿瘤类型,也是需要思考的问题。
总之,还是那句老生常谈的话,无论是药物开发还是临床治疗,都应该以患者为中心,都应该选择患者最合适、最需要的药物。