癌症基因组学研究最全综述回顾
来自Illumina 的一个英文癌症综述:《Cancer Research Review》

本文1.9万字,这篇综述涵盖了癌症研究的各个方面。文末推荐一本很不错的癌症科普书。
癌症,一种慢性的基因病。
简介
在癌症研究中,每个癌症样品呈现在研究人员眼前的已经是一个发生了改变的基因组,其中包含着独特且难以预测的诸多点突变、序列的插入缺失、易位、融合以及其他畸变。并且,这些发生的变异中,许多往往都是之前所未观察到的(Novel mutations),它们也不会只存在于基因组的编码区域中,因而为了能够真正做到全面研究癌症基因组本身所发生的所有突变事件,全基因组测序已被视为肿瘤基因变异研究中 唯一 严谨的方法。
然而,在所有这些变异中,却只有少数的几个主导着癌症这一疾病的发展和演变。要有效揭示这一演变和发展的过程,就需要监控基因表达水平上的变化,那么RNA-Seq便是用以确定这些遗传改变是否会影响疾病发展有用技术。但遗传改变有可能影响所有的细胞过程,包括染色质结构、DNA甲基化、RNA剪接异构体、RNA编辑和microRNA(miRNA)等。这就意味着,只有对所有这些独立的过程都进行检测和综合分析,才能在癌症研究中取得真正的突破。这些内容我们都将在下文一一展开。
当前基因组测序技术的一大特点是在于能够在很短的时间内并行测得数十亿(甚至数百亿)的独立序列片段——read,而每个read来源于单个的DNA分子。由此产生的数据我们可将其视为是对DNA分子的随机抽样,这反过来代表肿瘤样品中每个细胞的基因组的情况。这是我们解开癌症的原因和机制的一种强大工具。
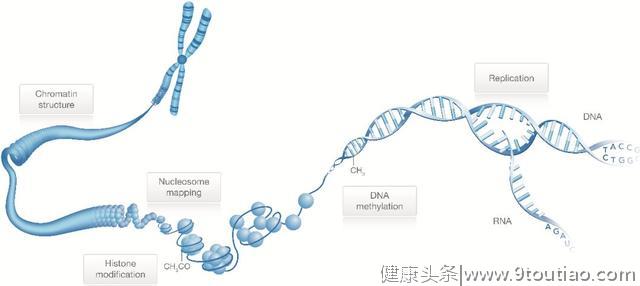
肿瘤异质性
癌症基因组的突变是复杂的。
每个人都携带一套独特的来自父母遗传的胚系突变(germline mutations)信息。但随着癌症的发展,体细胞突变(Somatic mutations)和基因组重排(genomics rearrangements)会逐渐增加。这些改变往往会引发耐药性以及转移。越来越多的研究证据表明,这些过程竟然可以是 有意的!——它们其实可以认为是癌细胞面临药物刺激的过程中不断进化的结果。我们想要全面地理解这一复发和耐药性的原因,就有必要进行纵向实验,按照癌症的发展过程,分阶段采集样品来进行研究。
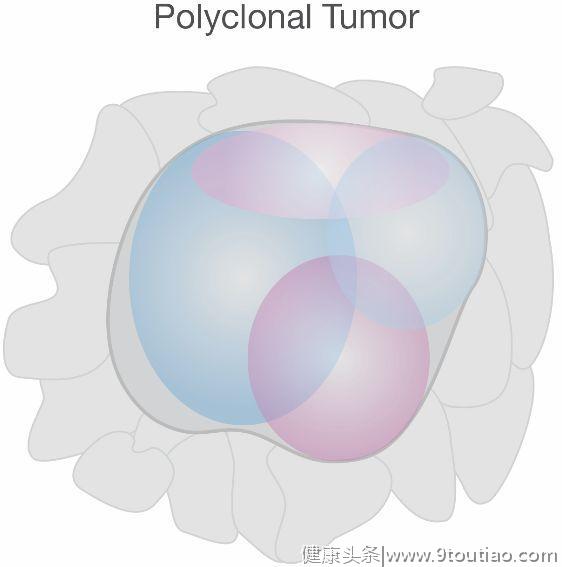
如上图,这是处于正常组织背景下的多克隆肿瘤(polyclone tumor)。大多数的肿瘤样品都会同时包含肿瘤细胞和正常细胞,如基质细胞、血管和免疫细胞。并且肿瘤本身也常常包含几种不同的克隆亚型(clone types),每一种都有着不同的治疗反应和复发可能。
根据传统病理学的估计,大部分研究的结果其实都只集中在那些肿瘤细胞比例 >60% 的区域中。并且为了确定哪些突变是肿瘤特异的,通常都要包含来自同一个体的正常组织样本作为参考。
然而,肿瘤本身也往往是异质性的。在癌症发展的过程中,个别细胞会发生新的突变,包含这些新突变的细胞会继续增殖,形成克隆亚型。因此,对于晚期癌症我们通常检测到的都是一个多克隆肿瘤,其中每一个克隆有一套独特的突变信息、独特的病理学和药物反应机制。这其实也正是癌症难以完全治疗的原因,而它的这种 异质性其实正是肿瘤基因组复杂性导致的一种表型性质。目前的深度测序可以检测样本中含量低至1%的克隆。
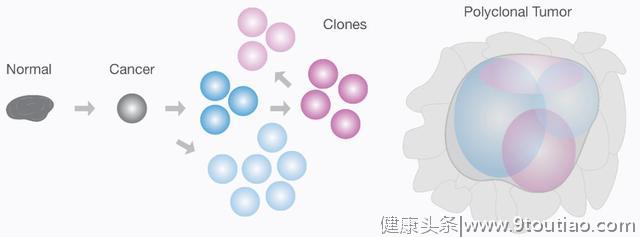
肿瘤内的异质性。体细胞突变的逐步累积产生了一个异质的多克隆肿瘤,其中不同克隆可能对治疗的反应不同。
而且,异质性一般还可以分两种情况讨论,第一种情况指的是:同一个病人的肿瘤细胞具有异质性。处于肿瘤发生的不同时期的肿瘤细胞的基因突变情况不同,造就了每一个肿瘤细胞群体内还有许多亚群(subclones),肿瘤细胞在通过转移时,就会有属于不同亚群的肿瘤细胞去侵入新的地方,形成新的肿瘤;第二种情况指的是:除了同一病人的不同肿瘤细胞会造成肿瘤的异质性外,肿瘤的异质性还体现在不同病人可能得了相同的肿瘤,但是那个『相同』未必真是相同——仅仅是表型相同,不代表着基因型也相同。
在某些基因中,突变频繁发生在同一个位置,这应该有特定的机制在起作用,这些有规律性的情况都会稍微容易对付。然而遗憾的是,对于大多数的基因,突变显然是随机出现在整个基因中的,这其实说明了DNA的复制和修复机制失灵了。

图中为两个假定的基因,他们有着两种不同的突变模式。深灰色框代表外显子组,而红色柱子代表存在突变的位置。A: 特定位置的频发突变可能表示有产生突变的生物学机制的参与。B: 散发突变存在于整个基因中,如P53,可能是由于复制和修复机制的失效。我们可以通过测序检测这两种情况下的突变。
参考文献
2012年Ding Li等人发表在Natrue上的一项研究弄清了急性髓细胞白血病(AML)的复发原因。他们发现了两个一般机理:(1)原发肿瘤中的始发克隆(founding clone)获得突变,进化成复发克隆;(2)始发克隆的一个亚克隆挺过了初次治疗,获得了更多突变并在复发时增殖。在一个病例中,原发肿瘤里原本仅占5.1%的亚克隆在复发后却成长为主要的克隆。而且对于所有的病例,化疗并不能够根除这些始发克隆。这项研究直接表明了在诊断后和初次治疗后检测并根除掉那些原本不起眼的小细胞群体有多么的重要!
Ding L., Ley T. J., Larson D. E., Miller C. A., Koboldt D. C., et al. (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481: 506-510
同样是2012年,同样是Ding Li这波人,同样是研究AML,他们这次发现大约三分之一的骨髓增生异常综合征患者会发展成继发性急性髓细胞白血病(AML)。这个研究的目的是为了确定骨髓增生异常综合征中的突变,它们有可能预测AML的进展。他们对七名继发性AML患者的七组皮肤和骨髓样品以及先前的骨髓增生异常综合征的配对骨髓样品一并做了全基因组测序。结果发现,在所有病例中,占主导地位的继发性AML克隆都是来自一个骨髓增生异常综合征的始发克隆。这其实就是说,骨髓增生异常综合征样品包含了对预后很重要的突变,针对这些突变的治疗方案是可能改善预后的。
Walter M. J., Shen D., Ding L., Shao J., Koboldt D. C., et al. (2012) Clonal architecture of secondary acute myeloid leukemia. N Engl J Med 366: 1090-1098
继续肿瘤治疗和预后的话题,Gerlinger M这一波人发起了一项研究,利用全外显子组测序来研究多个样品,这些样品不仅来自两名患者体内的原发肾癌,还包括了患者相关转移部位的空间分隔区域。最后,他们在原发肿瘤中看到了广泛存在的异质性现象!而且还特别指出了在每个肿瘤区域内有63%-69%的体细胞突变是无法检测到的。同时,还检测了同一肿瘤不同区域内预后良好和不良的基因表达特征。这其实还是突出了在突变积累之前早期诊断的重要性,以及对较大肿瘤需要进行多个部位的活检。利用同一名患者的多个样本得到的信息,他们能够重建疾病的发展进程!这是一种极为强大的方法,不仅能检测引发事件,还能检测表现出平行进化的基因。平行进化通常是基因在进化压力下的一种表现,其实也表示那些基因可能成为有效的治疗靶点。
Gerlinger M., Rowan A. J., Horswell S., Larkin J., Endesfelder D., et al. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883- 892。
转移
肿瘤的转移是一个复杂的过程,其中癌细胞脱离原发肿瘤,通过血液或者淋巴系统循环到身体的其他部位。在新部位,细胞继续繁殖,最终形成更多肿瘤,这些肿瘤包含了反映其组织来源的细胞。肿瘤(胰腺癌和葡萄膜肿瘤)转移的能力大大增加了它们的致死性。关于转移肿瘤的克隆结构、转移酶之间的系统发育关系、转移和原发部位的平行进化规模,肿瘤如何扩散,以及肿瘤微环境在转移部位决定中的作用如何等,许多这些基本问题目前仍然没有很好地解决。
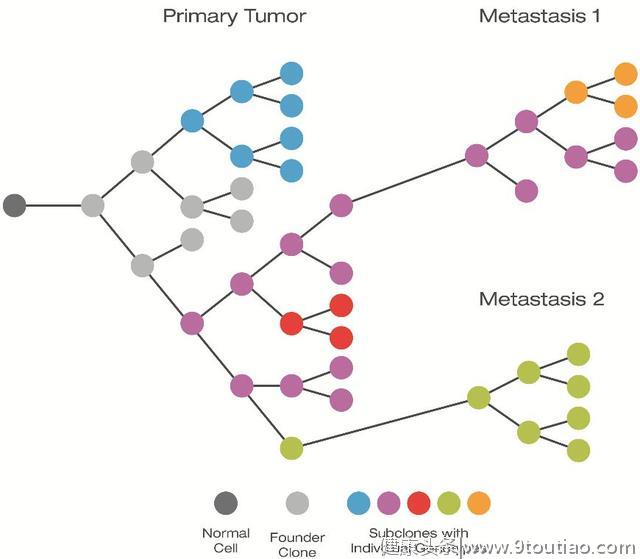
转移瘤可能来源于原发肿瘤中一个主要克隆(如上图:Metastasis1),也可能来源于次要克隆(Metastasis2)。转移瘤也会经历克隆进化(如Metastasis1所示)
参考文献
Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., et al. (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55-61
这篇文章证明了前列腺癌基因组经由致癌mTOR通路的特殊翻译机制,这其中产生了一个特定的基因群,它们参与了细胞增殖、代谢和侵袭。随后作者对一类翻译控制前侵袭信使RNA进行了功能鉴定,并指出了这些mRNA主导了癌症侵袭和转移。
基因组突变
所有的肿瘤在其发展的过程中都会不断积累体细胞突变(somatic mutations)。大多数常见的肿瘤与不同的癌基因相关联,这些癌基因以低频率发生突变。从大型癌症数据库中观察到的一个最令人惊讶的现象是癌症间甚至各个癌症类型内的显著遗传异质性。然而,似乎只有有限的细胞通路对肿瘤的细胞生物学很重要。目前很多人正在编辑收录各种癌症类型的体细胞突变综合列表,这对于更好地了解这种疾病背后的机制将有很大的指引作用。
研究参考
Nik-Zainal S., Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., et al. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979-993
这篇文章中研究了21个乳腺癌基因组,并给出了它的一个体细胞突变列表。发现带BRCA1或者BRCA2突变的癌症会有一种特别的替换突变特征和与众不同的缺失图谱。文章中还描述了一种局部的超突变现象,这称为『kataegis』(kataegis,希腊语中『雷雨』的意思,文中指的是在一个小区域中出现大量突变的机制,如下图)。并且这些区域中的碱基替换几乎都发生在TpC二核苷酸的胞嘧啶上!
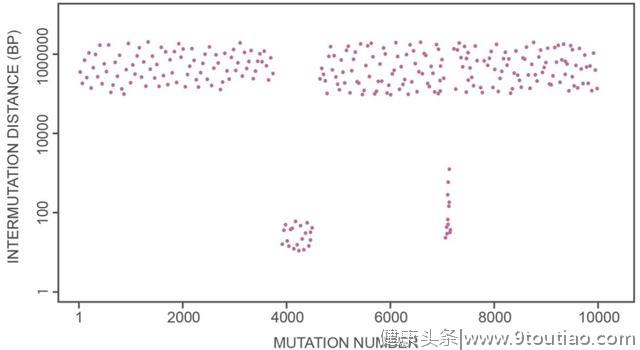
这是Kataegis图像。纵轴是突变间距(对数刻度)。这个图中基因组内大部分的突变都有着1Mbp左右的突变距离。其中超突变区即是表现为突变间距较低的簇。
Govindan R., Ding L., Griffith M., Subramanian J., Dees N. D., et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150: 1121-1134
这是另一篇文章,主要是对17个非小细胞肺癌(NSCLC)患者的肺癌及癌旁正常组织样本进行了全基因组和转录组测序。值得注意的是吸烟者中所观察到的突变频率比不吸烟者高10倍!这是通过深度测序揭示出的这些群体间所不同的克隆模式。而且其中所有经过验证的EGFR和KRAS突变都存在于原始克隆中,这其实也就表明了它们在癌症启动中可能发挥非常重要的作用。
镶嵌性
对于这个现象我们也同样通过实际的研究来说明。AML(急性髓细胞白血病)基因组中发现的大多数突变实际上是随机事件,在造血干细胞/祖细胞(HSPC)获得原始突变之前就存在了;但是随着克隆的扩增,细胞的突变历史被『捕获』了。如何理解这里的『捕获』?其实说的就是,原本那些突变都没什么鸟用,就摆在那无所事事,但是,在许多情况下,偏偏只需要再来一个或者两个额外的突变来协助就能共同作用,最后产生恶性的原始肿瘤克隆!
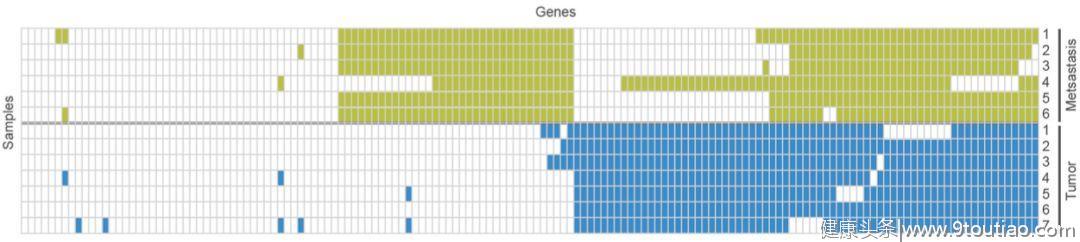
原发肿瘤和转移的镶嵌性。非同义点突变和插入缺失(绿色块)的区域分布的假设热图。行代表来自七个原发肿瘤区域和六个转移区域的样品。
研究参考
Abyzov A., Mariani J., Palejev D., Zhang Y., Haney M. S., et al. (2012) Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 492: 438-442
这里作者发现了一个现象:平均而言,iPSC细胞系表现出2个拷贝数变异(CNV),而这些CNV在iPSC来源的成纤维细胞中不明显。他们发现,至少50%的CNV以低频体细胞变异存在于亲本的成纤维细胞中。根据这一观察,他们估计大约30%的成纤维细胞的基因组中携带体细胞CNV,这表明体细胞镶嵌性广泛存在于人体中。
基因融合
基因融合是非常普遍的,也是癌症的一个重要特征。现在的研究发现,一个强启动子与一个下游功能基因(比如:原癌基因)的融合在某些癌症中很普遍。据估计,半数的前列腺癌含有TMPRSS2和ETS转录因子家族成员之间的融合。基因融合是由两个原本分开的基因或位点融合形成的。他们可能形成一种基因产物,很多时候表现出来的功能都是全新的,与两个融合的基因个体都不同。这种阴差阳错的情况可能引起致癌机制的激活,就像费城染色体阳性急性淋巴细胞白血病一样。这种基因融合导致BCR-ABL酪氨酸激酶表达,从而激活细胞增殖。有几种机制会导致基因融合的发生,这个现象是一些癌症类型的特点。胰腺癌的特点便是染色体重排的频繁断裂-融合-桥循环。目前有几种方法可以通过测序研究融合事件,如对肿瘤的全基因组测序和mRNA-Seq。
mRNA-Seq与全基因组测序组合的方法对于发现基因融合及其机制特别高效。原因就是mRNA-Seq可以提供直接的证据,来支持观察到的融合是否发生,并同时为融合基因是否表达提供了证据。而全基因组测序可以发现那些mRNA-Seq所发现不了的区域的信息,如基因间区和UTR。
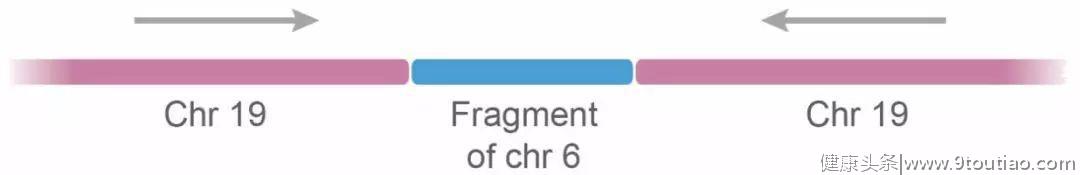
由折回倒位所引起的融合事件可捕获基因组中遥远区域的片段,如着丝粒重复或参与体细胞重排的区域。在这个例子中,6号染色体上的片段被插入到19号染色体上的重复区域之间。注意19号染色体的第二个拷贝是倒置的,这是折回倒位的特点。
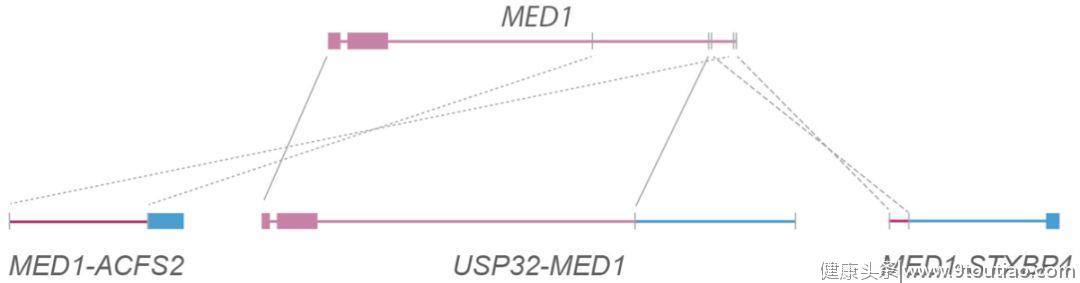
MED1(红色)与几个伙伴基因(蓝色):ACSF2,USP32 和 STXBP4形成基因融合。
实验上的设计参考
以Pair-end进行全基因组测序是目前检测基因融合最准确、最全面的工具,这些融合包括重复、倒位、通读和单碱基插入缺失。可以说Pair-end是检测融合基因成功与否的一个关键因素。
另外就是高深度测序结合更长的读长可以分辨融合连接中微同源的单碱基。而且这种能力是测序独有的。
研究参考
Robinson D. R., Wu Y. M., Kalyana-Sundaram S., Cao X., Lonigro R. J., et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 45: 180-185
文章主要是利用了全外显子组和转录组测序发现了转录抑制因子NAB2与转录激活因子STAT6的基因融合现象。其中27个独立性纤维性肿瘤(SFT)的转录组测序发现所有肿瘤中存在NAB2-STAT6基因融合。NAB2-STAT6基因融合的过表达诱导了培养细胞的增殖,并激活了EGR应答基因的表达,最后导致了肿瘤。
Seshagiri S., Stawiski E. W., Durinck S., Modrusan Z., Storm E. E., et al. Recurrent R- spondin fusions in colon cancer. Nature 488: 660-664
这一篇文章则主要分析了70个原发性人结肠癌的外显子组、转录组和拷贝数变异。拷贝数和RNA-Seq的数据分析确定了在一部分结直肠癌中存在IGF2的扩增和相应过表达。他们还利用RNA-Seq,在10%的结直肠癌中发现了与R-脊椎蛋白家族成员(RSPO2和RSPO3)相关的基因融合。这项研究表明了综合多项技术去了解复杂的癌症基因组很重要。
Thompson-Wicking K., Francis R. W., Stirnweiss A., Ferrari E., Welch M. D., et al. (2012) Novel BRD4-NUT fusion isoforms increase the pathogenic complexity in NUT midline carcinoma. Oncogene
这篇文章则提到了PER-624中一种新的BRD4-NUT融合竟然编码了一种功能蛋白,并且它对这些细胞的致癌机制很关键。BRD4-NUT融合转录本是通过易位后的RNA剪接而产生的,这似乎是这些癌症的一个共同特征。这种有助于融合基因的替代异构体表达的机制是第一次报道。
Wen H., Li Y., Malek S. N., Kim Y. C., Xu J., et al. (2012) New fusion transcripts identified in normal karyotype acute myeloid leukemia. PLoS ONE 7: e51203
在这项研究中,作者运用双端RNA-Seq来发现染色体核型中的融合,它们经传统的细胞遗传学分析未检测到异常。他们发现了临近基因间的融合转录本以及7个只存在于正常核型中的融合本。
染色体碎裂
这是一个不希望发生的现象,染色体碎裂是一个一次性的细胞危机,在单次事件中发生数十次至数百次基因组重排。这种灾难性事件的后果是复杂的局部重排和拷贝数变异,其中染色体上2个(偶尔3个)拷贝的有限范围可被检测。这种单次灾难性事件的模式不同于癌症发展的逐步积累突变的典型模式。在突变积累的癌症发展模式中,拷贝数无上限,因此通常有一个较大的范围。据估计,在所有癌症及其不同亚型之间,染色体碎裂的发生概率约2-3%,而在骨癌中发生概率则大约25%。
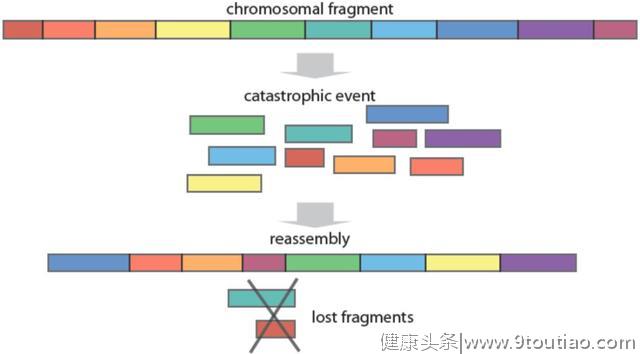
染色体碎裂的图示
研究参考
Rausch T., Jones D. T., Zapatka M., Stutz A. M., Zichner T., et al. (2012) Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements with TP53 Mutations. Cell 148: 59-71
文章提到一名Sonic-Hedgehong髓母细胞瘤(SHH-MB)患者的大量、复杂的染色体重排,此患者带有生殖细胞系TP53突变(Li-Fraumeni综合征)。同时将规模扩大到11名Li-Fraumeni综合征患者的筛查,发现有36%的肿瘤表现出与染色体碎裂一致的重排。这比一般肿瘤群体所观察到的2%染色体碎裂发生率要高得多。P53的生殖细胞系突变与一些肿瘤中凋亡中止导致染色体碎裂的假说是一致的。
拷贝数变异(CNV)
结构性变异影响基因量——可转录基因的功能拷贝数。肿瘤发展、药物反应及耐药性的发生通常是由基本的基因扩增和删除来驱动的。这些基因组上的改变可分成大的畸变和小的畸变。大的畸变包括整个染色体或部分染色体的丢失或重复,这被称为非整倍体。小的畸变可能只包含一个碱基,比如点突变和小片段的插入缺失。与健康的基因组不同,这些基因表达的改变会受到转录因子的严格调控,癌症基因组则通过基因的重复和删除来适应和逃避这种调控。癌症耐药性的发生正是此反应的速度和效率的绝佳证明。
基因表达
基因表达分析测定基因转录、RNA加工和表观遗传控制的产物。因此,基因表达分析不仅可以看出这些过程的『健康』程度,也可以深入研究细胞里面的分子机制。基于芯片的mRNA分析曾在癌症的基因变得研究中广泛使用,但基于测序的mRNA分析(mRNA-Seq)的出现代表我们测定和解析基因表达产物能力的又一次飞跃。mRNA-Seq可检测修饰过的RNA和表达水平极低的RNA的能力让它特别适合癌症研究。基于mRNA-Seq的方法也可检测非常快的转录变化、剪接异构体、融合基因以及可变聚腺苷酸化位点。

Feng H., Qin Z. and Zhang X. (2012) Opportunities and methods for studying alternative splicing in cancer with RNA-Seq. Cancer Lett
这篇综述关注了RNA-Seq在研究癌症相关的可变剪切中的应用。文中包含一个生物信息学工具列表,以及有关估计可变剪切异构体的表达水平的详尽讨论。
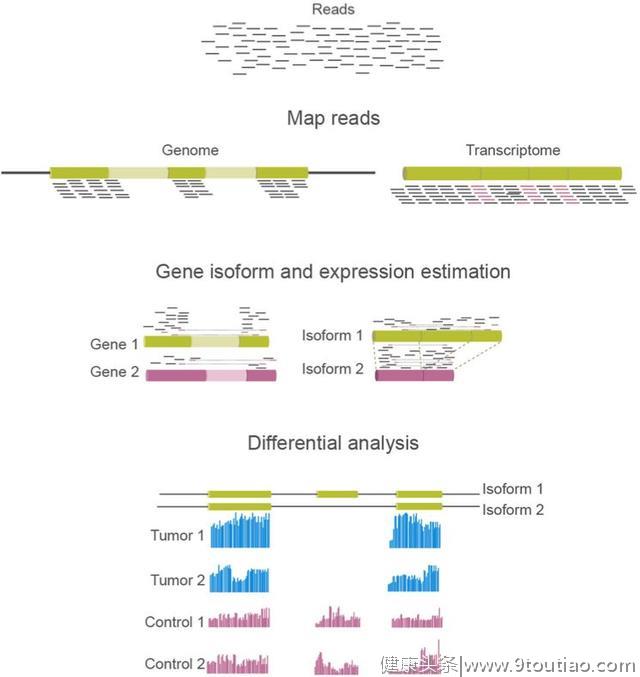
利用RNA-Seq研究癌症中基因表达和选择性剪接的典型生物信息学流程。首先,将短read定位到参考基因组或转录组。在定位之后,估算注释基因和转录本的表达与剪接。
van Delft J., Gaj S., Lienhard M., Albrecht M. W., Kirpiy A., et al. (2012) RNA-Seq provides new insights in the transcriptome responses induced by the carcinogen benzo[a]pyrene. Toxicol Sci 130: 427-439
作者发现,RNA-Seq所检测到的基因比芯片技术多约20%,而表达差异明显的基因更是接近三倍之多。因此,他们检测到的受影响的通路和生物学机制达2-5倍。作者还在许多基因中发现了可变异构体的表达,包括细胞死亡和DNA修复的调控因子,如TP53、BCL2和XPA,它们与基因毒性反应相关。他们还发现了功能未知的新亚型,如已知转录本的片段、带有额外外显子的转录本、内含子保留或外显子跳跃事件。
Kaur H., Mao S., Li Q., Sameni M., Krawetz S. A., et al. (2012) RNA-Seq of human breast ductal carcinoma in situ models reveals aldehyde dehydrogenase isoform 5A1 as a novel potential target. PLoS ONE 7: e50249
作者将三个DCIS模型(MCF10.DCIS、SUM102和SUM225)的表达与三维(3D)覆盖培养的非致癌乳腺上皮细胞的MCF10A模型进行了比较,确定了DCIS模型共用的表达变化。他们发现,差异表达的基因编码了与多个信号通路相关的蛋白。
Meyer J. A., Wang J., Hogan L. E., Yang J. J., Dandekar S., et al. (2013) Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45: 290-294
作者利用RNA测序,报道了诊断和复发相配对的骨髓标本的转录本图谱,这些标本来自十名患有小儿B淋巴细胞白血病的个体。转录组测序鉴定出20个新获得的突变,它们不存在于最初的诊断中,而2名个体带有复发特异的突变。带有NT52C2突变的所有个体都在初步诊断后36个月内复发。
实验设计上的注意事项
RNA-Seq已成为一种研究肿瘤分子变化的常规应用,大部分研究人员采用生产商的试验流程。rRNA的去除可提高信噪比,实现低表达转录本的检测。
癌症中的体细胞突变基本上是 de novo。测序不需要关于突变的先验知识,即可准确定位突变以及得到转录本丰度。
肿瘤通常包含各种细胞。mRNA-Seq的可延伸检测范围和准确性对检测微小的表达变化非常宝贵。只要肿瘤转录本包含了独特的体细胞突变或剪接变异体,那么就可将它与正常的细胞区分开来。
二代双端对读测序检测基因融合的灵敏度取决于许多因素,包括表达水平、转录本长度、所使用的样品制备方法以及cDNA文库的片段长度。
大部分实验方案采用poly(A)富集的RNA制备方法来测定mRNA水平。然而,非编码RNA,如miRNA,也在细胞的生物学中发挥重要作用,并常常介导对肿瘤生长和存活很关键的过程。非编码RNA可通过现有的poly(A)-(rRNA去除)实验方案轻松分析。
RNA表达是组织和细胞类型特异的。在选择肿瘤-正常对照中的对照时,应考虑这一点。
选择性剪接
癌症的生物起源、发展、转移与转录组中的许多变异相关联。癌症特异的选择性剪接是个普遍存在的现象,也是个主要的转录后调控机制,涉及到许多癌症类型。
Seo J. S., Ju Y. S., Lee W. C., Shin J. Y., Lee J. K., et al. (2012) The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res 22: 2109-2119
作者分析了韩国200个肺腺癌。他们在LMTK2、ARID1A、NOTCH2和SMARCA4中发现了新的驱动突变。他们还发现了45个融合基因,其中8个是嵌合的络氨酸激酶。在17个反复发生的选择性剪接事件中,原癌基因MET中的第14号外显子跳过可能是癌症驱动因素。这项研究表明了这种癌症的复杂性以及运用几种技术的价值。
Liu J., Lee W., Jiang Z., Chen Z., Jhunjhunwala S., et al. (2012) Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res 22: 2315- 2327
作者对19个肺癌细胞系和3组肺部肿瘤/正常样本配对开展了全基因组测序和转录组测序。他们鉴定出106个与癌症特异性的异常剪接相关的剪接位点突变,包括一些已知的癌症相关基因中的突变。RAC1b是RAC1 GTP酶的一个异构体,含有一个额外的外显子,被认为在肺癌中优先上调,并对MAP2K(MEK)抑制剂PD-0325901敏感。
Thompson-Wicking K., Francis R. W., Stirnweiss A., Ferrari E., Welch M. D., et al. (2012) Novel BRD4-NUT fusion isoforms increase the pathogenic complexity in NUT midline carcinoma. Oncogene
这篇文章发现了PER-624中一种新的BRD4-NUT基因融合编码了一种功能蛋白,它对这些细胞的致癌机制很关键。BRD4-NUT融合转录本是通过易位后的RNA剪接而产生的,这似乎是这些癌症的一个共同特征。这种现象以及促进融合基因的可变异构体表达的机制,过去一直未被发现。
RNA编辑
在我们人体中,DNA和RNA序列之间的差异也被称之为 RNA编辑,这是一种广泛存在的现象。最频繁的RNA编辑类型是通过腺苷脱氨酶作用于RNA(ADAR)从而实现由腺苷到肌苷的转换。然后紧接着,剪接和翻译机制会将肌识别为鸟苷。在一些肿瘤基因组比正常基因组有着更更比例的RNA-DNA差异。
Jiang Q., Crews L. A., Barrett C. L., Chun H. J., Court A. C., et al. (2013) ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A 110: 1041-1046
作者发现,慢性髓细胞白血病(CML)急变期的祖细胞有着更高的IFN-r通路基因表达以及BCR-ABL扩增。在CML发展期间,他们还发现IFN应答的ADAR1 p150亚型的表达增强,并且腺苷-肌苷的RNA编辑增加。
MicroRNA和非编码RNA
MicroRNA(miRNA)的长度很短,大小集中在17bp-25bp之间,属于非编码RNA(ncRNA)家族的成员。它们调控多种不同的生物学功能,包括发育、细胞增殖、细胞分化、信号转导、凋亡、代谢和细胞寿命。
通过RNA-诱导沉默复合物(RISC)与转录本的3-UTR或者与编码区中的识别位点相互作用。miRNA的一个主要作用是抑制基因的转录后表达。在多种癌症中,许多miRNA位于存在序列缺失删除或扩增的基因组区域,这表明它们很可能在癌症的发展过程中扮演很重要的角色。miRNA的编辑位点已经在近年来的研究中被发现了,表明RNA编辑和miRNA介导的调控之间可能存在着关联。同时由于miRNA的测定简单、相对稳定,并且在大量mRNA的控制上起作用,这就让miRNA成为癌症诊断以及治疗期间的检测和分期过程中极具吸引力的标志物。
Law P. T., Qin H., Ching A. K., Lai K. P., Co N. N., et al. (2013) Deep sequencing of small RNA transcriptome reveals novel non-coding RNAs in hepatocellular carcinoma. J Hepatol
这篇文章描述了一种新的PIWI-互作RNA(piRNA)piR-Hep1,它参与了肝脏肿瘤发展。与周围正常肝脏相比,46.6%的肝癌细胞(HCC)存在piR-Hep1表达上调。piR-Hep1的沉默抑制了细胞活力、运动和侵袭。作者还发现miR-1323在HCC中的大量表达,以及它与肝硬化背景下所产生的肿瘤的独特关联。
实验设计上的注意事项
测序深度与检测灵敏度是直接相关的。在典型的实验中,如果测序流动槽(Flowcell)的一条通道(lane)只上一个样本,那么测序深度会非常高,这能实现极其灵敏的检测。基于此原因,miRNA的测序深度很少成为考虑因素。在筛查应用中,或不需要如此高深度检测的研究中,样品可加上测序Index标签,从而能够在同一个测序lane中上样多个不同的样本。需要注意的是,在设计miRNA的测序深度时,要时刻记住miRNA控制基因表达,因而miRNA水平的小变化可能影响许多编码蛋白。
新发现的miRNA应当通过功能分析(如Ago2结合或敲除实验)来确认。
实验应当包含足够的样品,以便结论具有真正的统计意义和高的可信度。一般来说,建立一个可能的假设相对来说是比较容易的,只要能证明miRNA存在于患者的肿瘤中,即便样本数量不多也是足够的。但是要真正验证假设就没那么容易了,通常需要大量的患者来检验,并建立统计学可信度。目前,关于如何在测序研究中建立统计学可信度和多重检验纠正,还没有特别公认的方法。当前基于测序的miRNA分析并没有实现miRNA表达的绝对定量,而仅是不同miRNA(如肿瘤-正常配对)的相对量。
样品分层是癌症样品的一个问题。一个特定的癌症表型可能代表几种不同的病因和机理。为了要进行严格的分析,应当有足够多的样品量,从而能够充分代表每个肿瘤亚型。而miRNA的表达会随着肿瘤的发展而变化,因此在实验设计时应建立肿瘤分期和分级。
RNA-蛋白结合(CLIP-Seq)
在人类细胞中,大多数mRNA(或前体mRNA)与核不均一性核糖蛋白(hnRNP)相结合,形成大的hnRNP-RNA复合物。hnRNA蛋白在RNA加工的所有关键环节中都发挥重要作用,包括前体mRNA剪接以及mRNA出核、定位、翻译和稳定性。几十种RNA结合蛋白(RBP)和基因的hnRNP蛋白与癌症相关联。
RNA-蛋白的相互作用可通过交联免疫沉淀测序(CLIP-Seq)来测定。在CLIP-Seq中,细胞经过紫外线处理,让RBP与RNA复合物共价交联。细胞随后被裂解,RBP-RNA复合物被免疫共沉淀,从而测序相应的RNA。
Wilbert M. L., Huelga S. C., Kapeli K., Stark T. J., Liang T. Y., et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell 48: 195-206
LIN28是个保守的RNA结合蛋白,它与多能性、重编程和肿瘤的形成相关。在各种不同的癌细胞和原发肿瘤组织中都发现LIN28的异常上调。在这篇文章中,作者利用CLIP-Seq鉴定了大约四分之一分布于人类转录本中LIN28的结合位点。从这些结合位点中他们发现,LIN28与mRNA中富含环结构的GGAGA序列结合。同时还发现了,LIN28的表达能够导致可变剪切中的下游变化。
表观遗传和甲基化
癌症发展过程中的表观遗传改变与异常的基因表达相关联。近期的研究证据表明,表观遗传上的改变可能在癌症 起始中 发挥作用。表观遗传的控制是通过多个不同过程进行介导的,包括DNA修饰(甲基化或乙酰化)、组蛋白修饰和核小体重塑。发现控制表观基因组的基因发生突变,在人类癌症中是一个相当普遍的现象。NGS测序技术提供了一整套工具,可定位突变并测定它们对癌症发展的影响。
癌症中表观遗传修饰物的基因突变。在不同类型的癌症中经常观察到三类表观遗传修饰物发生突变,这突出了遗传学和表观遗传学之间的串扰。表观遗传修饰物的突变有可能引起癌症的全基因组表观遗传改变。了解遗传和表观遗传变化的关系将为癌症治疗提供新的见解。
DNA修饰
DNA修饰目前可以很容易地通过多种技术来进行测定。不同技术的选择取决于所需的通量和分辨率。

Bert S. A., Robinson M. D., Strbenac D., Statham A. L., Song J. Z., et al. Regional activation of the cancer genome by long-range epigenetic remodeling. Cancer Cell 23: 9-22
文章作者通过协同的长距离表观遗传激活(LREA)技术,鉴定出一种结构域基因去调控的机制。这些区域通常会跨越1Mb的基因组长度,包括关键癌基因、microRNA和癌症的生物标志物基因。LREA结构域中基因启动子的特点是活性染色体标记的的获得和抑制标记的丢失。
Brastianos P. K., Horowitz P. M., Santagata S., Jones R. T., McKenna A., et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 45: 285-289
为了鉴定和验证脑膜瘤中的体细胞遗传改变,作者对17个脑膜瘤进行了全基因组或外显子组测序,并对另外48个肿瘤进行了靶向测序。他们所观察到的突变谱是分布广泛,但他们证实了43%的肿瘤中存在病灶NF2失活,并在另外8%的肿瘤中发现表观遗传修饰物的改变。
Duncan C. G., Barwick B. G., Jin G., Rago C., Kapoor-Vazirani P., et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res 22: 2339-2355
脑胶质瘤、急性骨髓性白血病和软骨瘤中经常发生NADP+依赖的异柠檬酸脱氢酶IDH1和IDH2的单等位点基因点突变。作者表明,IDH1R132H等位基因的杂合表达足以诱导这些肿瘤特有的以DNA甲基化为特征的全基因组改变。这说明了IDH1R132H/WT突变体是推动人类癌细胞的表观遗传不稳定性的原因。
Zhang J., Benavente C. A., McEvoy J., Flores-Otero J., Ding L., et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481: 329-334
视网膜母细胞瘤是一种发生于视网膜的侵袭性儿童癌症,由RB1失活所引发,但潜在机理仍然未知。在此类高度侵袭性的癌症中,许多基因都参与其中,但RB1是唯一的已知发生突变的癌基因。与有限的体细胞突变不同,相对正常的成视网膜细胞,肿瘤的甲基化图谱表现出巨大的改变。最惊人的结果之一是人视网膜母细胞瘤中原癌基因脾络氨酸激酶(SYK)的诱导表达。SYK是肿瘤细胞生存所必需的。研究人员接着表明,小分子抑制剂对SYK的抑制导致培养和体内的视网膜母细胞瘤的细胞死亡。
实验设计上的注意事项
每种组织和细胞类型都有着独特的甲基化模式;因此必须获得感兴趣的组织以便分析。癌症研究中,肿瘤组织-癌旁正常组织的配对可简化分析。
通过重亚硝酸氢盐测序所产生的超大量CpG标志物很难解释,且可靠的统计学分析目前仍然困难重重。不过,下面一些实际的方法可简化分析:
- RRBS-Seq通过限制覆盖度而简化分析;
- 综合分析大大改善了结果的可解释性。例如,将表达分析与甲基化分析相结合,让我们可专注于表达水平改变的基因;
- 将分析限制在某个感兴趣的基因或区域中。这种方法对GWAS的后续研究很有效,也适合已有实验证据说明目的区域存在基因调控或染色质重塑的研究。与降低代表性的方法不同。这种方法实现了更多区域的分析,因此可获得更多信息。
组织培养物应当谨慎使用。随着时间的推移和组织增殖,培养物的甲基化水平可能已经改变,不大能代表原先的组织样本。
组蛋白修饰
组蛋白修饰通常指的是甲基化和乙酰化。组蛋白H3K9、H3K27和H4K20的甲基化与基因转录的抑制相关,而H3K4和H3K36的三甲基化与活性转录的染色质相关。组蛋白乙酰化几乎总是与染色质可接近性和转录活性水平的增高相关。通过操控染色质状态和DNA可接近性,表观遗传修饰在各个发育阶段、组织类型和疾病中都对基因表达的控制起着关键作用。
Wilkinson A. C., Ballabio E., Geng H., North P., Tapia M., et al. (2013) RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep 3: 116-127
这篇文章报道了一种转化机制,其中两个致癌融合蛋白合作激活目的基因,然后调节其下游产物的功能。
实验设计上的注意事项
每种组织和细胞类型都有着独特的甲基化模式;因此必须获得目的组织以便分析。
组蛋白修饰可以通过各种ChIP-Seq方法进行检测,原理是通过抗体与目标甲基化组蛋白进行特异结合。
同样,组织培养物应当谨慎使用。随着时间的推移和组织增殖,培养物的甲基化水平可能已经改变,不大能代表原先的组织样本。
染色质结构与重排
染色体重排需要DNA双链断裂的形成和连接。这些事件的发生,会破坏基因组的完整性,并经常在白血病、淋巴瘤和肉瘤中观察到。并且特定基因间的反复的基因融合在不同的个体中均观察到,这表明这些基因一定在细胞周期中的某个阶段他们之间的物理位置非常接近。
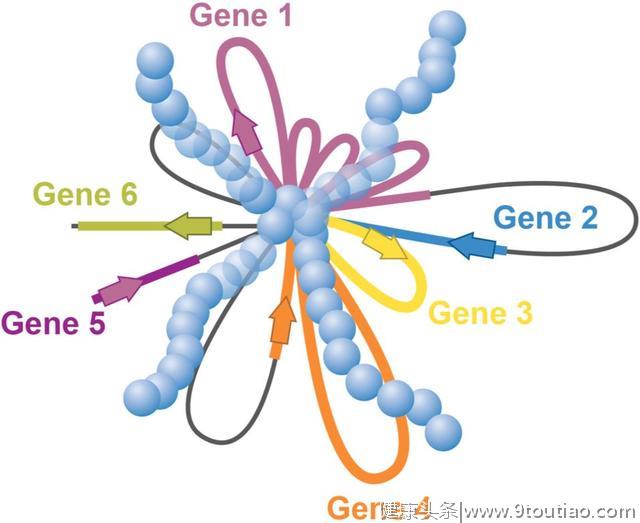
这是一个设想的三维、具有转录活性的复合物,它包含了致密的环化位置。这个示意图是基于检测到的成环事件,并假设所有成环事件都能发生在单个细胞内。在这个模型中,所有小环汇集到一个共同的核心(蓝色球)。环降低了转录活性复合物的物理大小,从而推动转录因子接近待定的基因组位点。
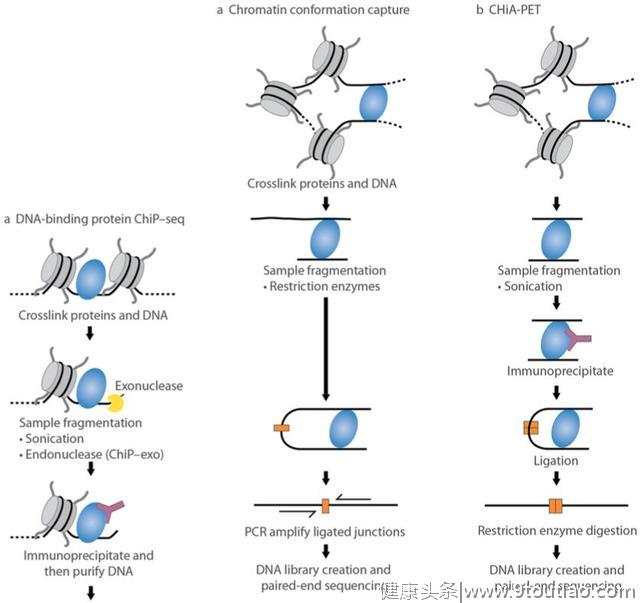
检测染色质相互作用。在三维空间中,相同或不同染色体上远端的基因组区域相互作用,而这种相互作用是由一个或多个DNA结合蛋白介导的。a) ChIP-Seq 利用染色质免疫共沉淀来鉴定DNA和蛋白的相互作用。人们采用各种DNA-片段化方法和核酸外切酶,来缩小片段的大小分布。b)染色质构像捕获实验利用一个连接步骤将互作的染色质片段相连接。这种方法可鉴定与遥远序列相结合的蛋白。c)配对末端标签测序分析染色质相互作用(ChIA-PET)同样也利用连接步骤来检测染色质相互作用,将不相邻的互作区域配对。然而,ChIA-PET利用染色质免疫共沉淀(ChIP)步骤,只能鉴定特定蛋白的相互作用,如RNA聚合酶II。
参考文献
Papantonis A., Kohro T., Baboo S., Larkin J. D., Deng B., et al. TNFalpha signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J 31: 4404- 4414
作者利用测序以及染色体构像捕获(3C)和ChIA-PET表明,TNF_alpha诱导响应基因聚集在分散的『NF-B工厂』。一些『工厂』还专门转录编码miRNA的响应基因,这些miRNA靶定下调的mRNA。
Rocha P. P., Micsinai M., Kim J. R., Hewitt S. L., Souza P. P., et al. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol Cell 47: 873-885
细胞核组织可决定『脱靶』活性以及融合伴侣的选择。这项研究表明,绝大多数已知的活化诱导胞嘧啶核苷脱氨酶(AID)介导的Igh转位伴侣在类型转换过程中与此位点接触的染色体结构中被发现。此外,这些相互作用结构域可用来鉴定被AID靶定的其他基因。
Theodoratou E., Montazeri Z., Hawken S., Allum G. C., Gong J., et al. Systematic meta- analyses and field synopsis of genetic association studies in colorectal cancer. J Natl Cancer Inst 104: 1433-1457
这篇研究文章表明,T细胞特异的转录因子GATA3在介导增强子接近调控区域上扮演了重要角色,这些调控区域参与了雌激素受体(ESR1)介导的转录。GATA3沉默导致在雌激素刺激之前辅助因子和活性组蛋白标记的整体重新分配。
Hakim O., Resch W., Yamane A., Klein I., Kieffer-Kwon K. R., et al. (2012) DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484: 69-74
作者发现,在培养的小鼠B淋巴细胞中,缺乏经常性的DNA损伤时,Igh或Myc与其他所有基因之间的易位与它们的接触频率直接相关。反过来,与经常性位点指向的DNA损伤相关的易位与DNA断裂形成的速率成正比。他们认为,非定向重排反映了细胞核结构,而DNA断裂形成决定了包括驱动B细胞恶性肿瘤在内的经常性易位的位置和频率。
综合分析(多组学分析)
所有的生物过程都是相互关联的,而在癌细胞发生过程的任何一个变化都会影响其他所有过程。突变可能影响所表达的活性,继而又影响DNA甲基化,再就影响其他许多基因的表达等等一连串的反应。每个个体都有着大量的特有突变,再加上这一连串的事件,能够让人们深入研究各种用于区分癌症的疾病表型。综合分析可以使揭示癌症生物学的真正复杂性向前迈进了一步。研究人员如今能够检测大部分的单个过程,但认识和治疗癌症的真正进步将来自于对所有这些过程的综合分析,也就是常说的多组学分析。
参考文献
Weischenfeldt J., Simon R., Feuerbach L., Schlangen K., Weichenhan D., et al. (2013) Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell 23: 159-170
作者发现,早发性前列腺癌的形成涉及到雄激素驱动的结构重排。相比之下,老年发病的前列腺癌积累了非雄激素相关的结构重排,表明一种不同的肿瘤形成机制。
Cowper-Sal lari R., Zhang X., Wright J. B., Bailey S. D., Cole M. D., et al. (2012) Breast cancer risk- associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet 44: 1191-1198
作者表明,与乳腺癌风险相关的SNP集中在FOXA1和ESR1的顺反组(cistrome),以及组蛋白H3赖氨酸4单甲基化(H3K4me1)的表观基因组。大多数的风险相关SNP调控FOXA1在远端调控元件的染色质亲和力,这导致等位基因特异的基因表达。
Peifer M., Fernandez-Cuesta L., Sos M. L., George J., Seidel D., et al. (2012) Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet 44: 1104-1110
作者发现了TP53和RB1失活的证据,并在编码组蛋白修饰物的基因中发现了反复的突变。此外,他们还在PTEN、SLIT2和EPHA7中观察到突变,以及FGFR1络氨酸激酶基因的病灶扩增。这种综合分析表明,组蛋白修饰可能与小细胞肺癌(SCLC)有关。
技术参考
一个良好的实验设计可以提高技术性能,从而产生最易解释和可靠的结果。这里重点强调研究人员在设计实验时必须牢记的生物学和技术的特性。
癌症中的实验设计面临一些独特的挑战。典型的肿瘤样本包含两个基因组:遗传自父母的生殖细胞系(germline)和在疾病发展过程中积累的体细胞突变(somatic mutations)。肿瘤细胞在样本中的比例可能在10%-100%之间。肿瘤基因组也是动态的,会快速积累 de novo 突变。因此,每一个肿瘤中又可能同时包含几个克隆亚型。
目前大部分已发表研究的样本量都非常小,可被视为仅是提出了相关的假说。而随着越来越多的测序信息被我们所获得,大部分癌症类型可根据其分子表型,被分成多个亚群。这严重降低了实验的能力,并增加了严格分析所需的样本数量。部分解决这一难题的方案是在探索阶段利用全基因组测序来寻找新的突变。在第二阶段,利用全外显子组或靶向测序来确认新发现的这些突变,并确定他们在大型队列中的丰度。然而,在未来,统计学上严谨的全基因组测序实验有可能是非常大的,需要数千个样本。
使用NGS测序技术进行深度测序是指多次生成定位到同一区域的序列片段,有时达上百次。但由于每条序列片段是从单个DNA分子中产生的,故提高深度测序能够实现原始样品中低至1%的克隆的检测。比较同一个体的肿瘤和癌旁正常组织的序列,我们可以很容易识别浸润组织的序列片段。最佳的读取深度将取决于癌症类型和所需的灵敏度,但一般建议为正常基因组最低为40倍的覆盖深度,而癌症基因组需要80倍以上的覆盖深度。在肿瘤高度异质时,可能需要肿瘤不同部位的多次活检,才能包含所有的细胞类型。
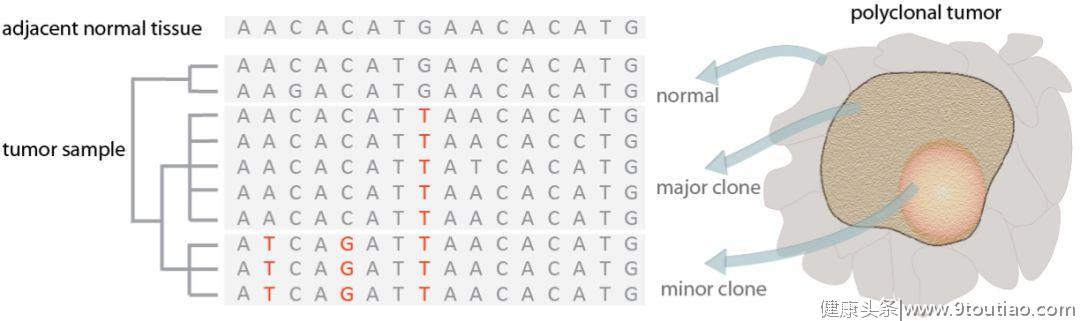
在这个假定的例子中,肿瘤含有两个癌症克隆和邻近组织。肿瘤样本中正常细胞所产生的序列(图中:比对中的前两个序列)可通过与邻近正常组织所产生的序列比较后确定。肿瘤样本中的剩余序列可分为两组,分别代表主要和次要的肿瘤克隆。次要克隆,若不及时治疗,可能在复发时成为肿瘤的主要组分。在实际分析中,肿瘤样本至少要有40倍的覆盖深度,并覆盖靶定的基因集合、全外显子组或全基因组。
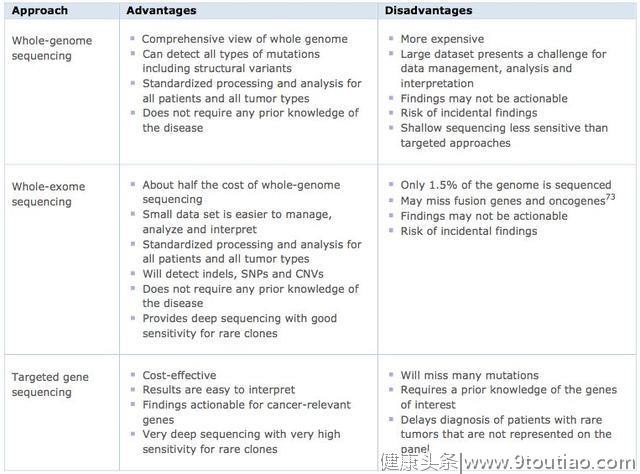
检测癌症基因组中的体细胞突变通常有三种方法:全基因组测序、全外显子组测序和靶向基因测序。下表简要介绍了每种方法的有点和缺点。在比较多发性骨髓瘤的全基因组和外显子组测序时,半数的蛋白编码突变通过染色体畸变(如易位)而存在,其中大部分不能单独被外显子组测序而发现。靶向重测序是一种有用的技术,可收录超大队列中已知癌症相关基因的突变。从长远来看,随着我们对基因组的了解日益加深,并且我们处理和解释大型数据集的能力的提高,全基因组测序无疑是最好的肿瘤分子鉴定方法。而在短期内,靶向基因测序可为患者匹配出市场上已有的药物,让他们立刻受益。
参考来源
"Illumina cancer research